
 分享
分享

今日与大家汇报原发性纤毛运动障碍(PCD)的进展,我认为对我们未来的工作有所帮助和启发。
一、PCD的定义及诊断流程
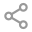
首先看PCD的定义,它强调以支气管扩张为主的呼吸道表现。它是因为先天性的纤毛结构或功能异常,或者多运动纤毛生成减少,导致的呼吸道黏液清除能力的障碍。
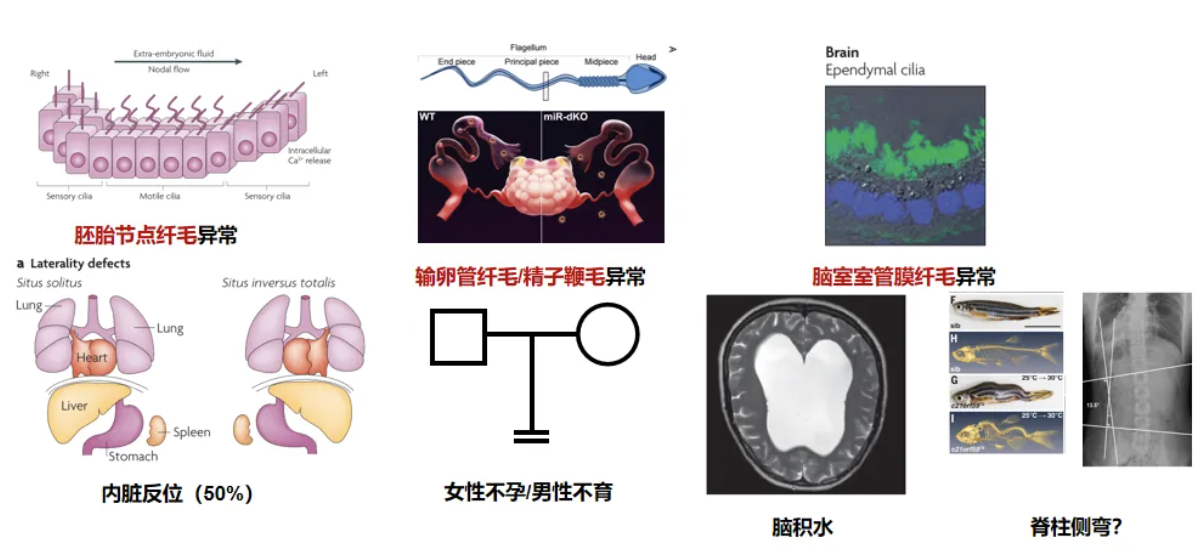
在临床上,如果患者符合以下特征:足月儿发生新生儿呼吸窘迫,早发的支气管扩张,反复不愈的鼻窦炎等症状,就应该考虑进行PCD相关的检查。在询问病史时,以呼吸病史为主,兼顾其他系统。同时应注重询问家族史,家系中有无类似情况。
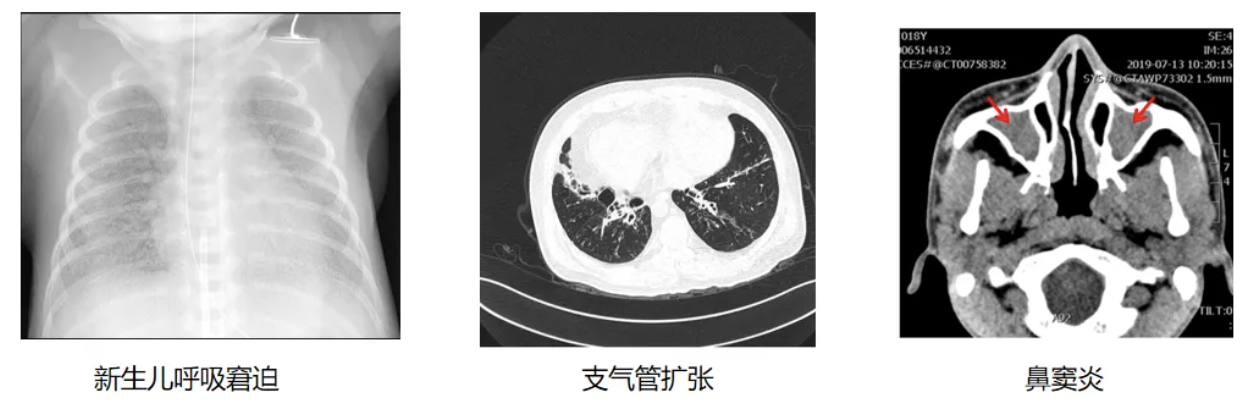
针对PCD高危人群的筛查实践,欧美国家已建立较为完善的体系。左图为美国胸科协会(ATS)诊断流程,其基于nNO检测、基因检测、透射电镜制定了系统性筛查方案;欧洲呼吸学会(ERS)同样具备标准化的诊断流程筛查机制,其额外强调高速视频显微分析(HSVA)在PCD诊断中的重要性。在我国目前主要遵循欧洲呼吸学会(ERS)指南开展筛查工作[1-2]。
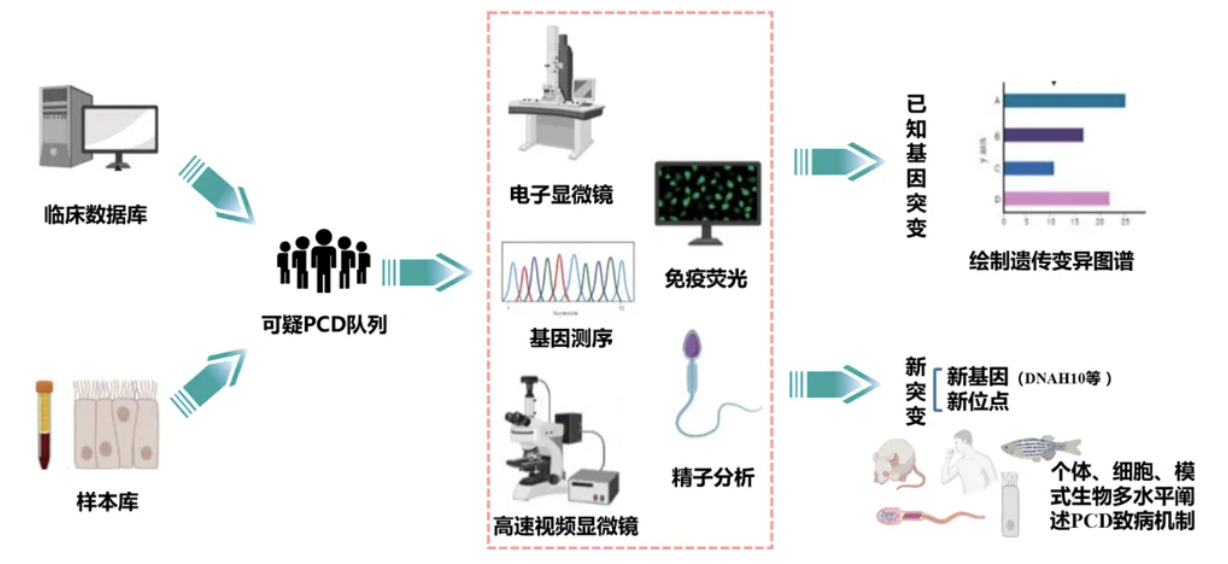
针对可疑人群的筛查,本团队致力于制定适用于我国的PCD诊疗流程,并旨在通过多种手段检测已知基因的突变,进而绘制遗传变异图谱。同时,我们可发现新基因及新位点;例如,本团队已识别出若干新基因与新位点。为证实新位点确属新发现,需通过个体细胞模型的多层次分析,阐明PCD是否表现其机制,并通过多个家系进行验证。鉴于原发性纤毛运动障碍大多为常染色体隐性遗传的单基因疾病,故围绕其作为遗传病的特性,尽管遗传病常被视为先天性疾病,但后天因素亦可引发基因改变。
二、PCD的诊断/常用诊断方法及基因检测
关于PCD的诊断方法,主要围绕“先天性”和“纤毛”:① 是否为“先天性”:基因检测,最常用的检测方法为全外显子测序。其他提示性信息:父母为近亲结婚、有支扩家族史(兄弟姐妹)、自幼起病;②“纤毛”相关检测:纤毛结构检测:纤毛透射电子显微镜、纤毛免疫荧光检测(纤毛结构蛋白的抗体)。纤毛功能检测:高速视频显微镜分析。呼吸道纤毛功能的间接体现:内脏反位、鼻窦炎、中耳炎、不孕不育、精液常规、精子透射电子显微镜;③ 经鼻一氧化氮检测(目前发现PCD和囊性纤维化会出现特异性下降,但机制不明)。欧洲呼吸协会和美国胸科协会的PCD确诊标准为:基因检测显示PCD双等位致病变异或者纤毛透射电子显微镜显示特异性的纤毛相关缺陷[3-4]。
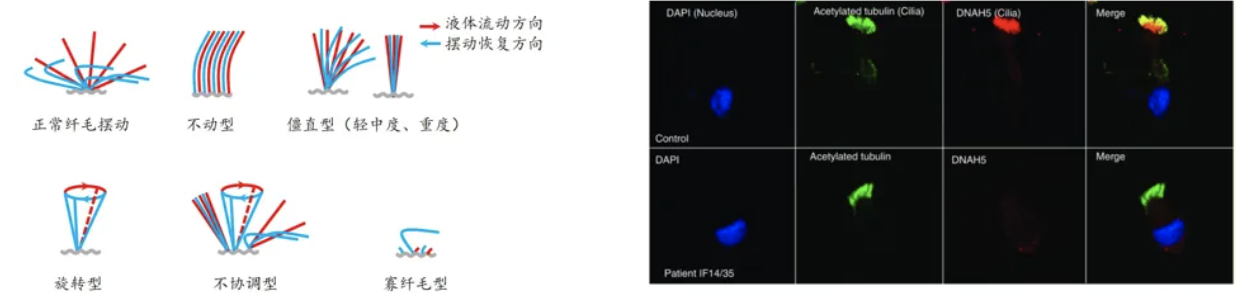
PCD常用诊断方法:1.鼻呼气一氧化氮检测(nNO)——指南优先。对于正常人,nNO检测结果通常大于77nL/min,研究表明PCD患者的nNO结果会出现特异性下降,但机制不明。需要排除技术误差和嗜酸性肉芽肿性多血管炎(EGPA)导致的慢性鼻窦炎和鼻息肉,至少间隔一个月检测2次。部分PCD患者nNO正常,所以nNO通常只作为PCD筛查的辅助手段,并不具有排除诊断的意义;2.高速视频显微镜(HSVA)——指南优先。直接取患者的气道纤毛细胞在显微镜下观察其摆动,通过计算纤毛摆动模式和纤毛摆动频率,直观的反映纤毛运动障碍。但仪器价格高,操作难度大;3.免疫荧光分析。取气道纤毛后通过免疫荧光染色判断纤毛结构是否完整[5]。
阳性可以确诊PCD,但阴性无法排除PCD。PCD的基因型比较多(超过50种),基因检测优先推荐全外显子测序(可检测所有基因的外显子变异)。二代测序(高通量测序)的结果需要再次进行一代测序(金标准)进行验证[6]。
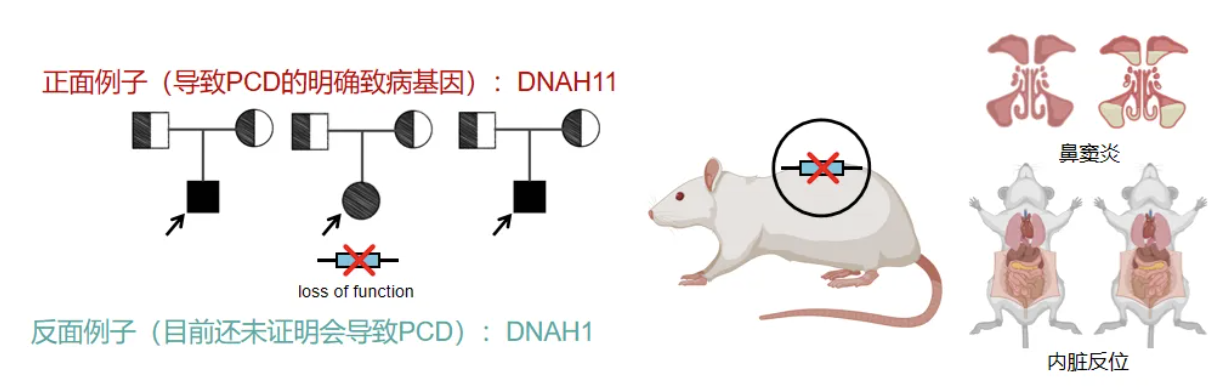
何为阳性结果?①基因的致病性明确(目前已有超过50个明确可导致PCD的致病基因)。需满足以下两个条件:既往有多个PCD家系报道该基因变异(变异的类型为明确loss of function的变异;动物实验成功验证基因变异会导致PCD。下面通过两个例子说明,我们在三个家系中发现DNAH11的纯合突变可以导致PCD的经典临床表现,且动物模型同样具有鼻窦炎和内脏反位,因此DNAH11被认定为是PCD的致病基因,而DNAH1既往只有一个DNAH1 loss of function报道的PCD家系,其他的都是错义变异或者非PCD家系。既往动物模型没有鼻窦炎和内脏反位的报道[7-8],因此目前不能认定它是PCD的致病基因。
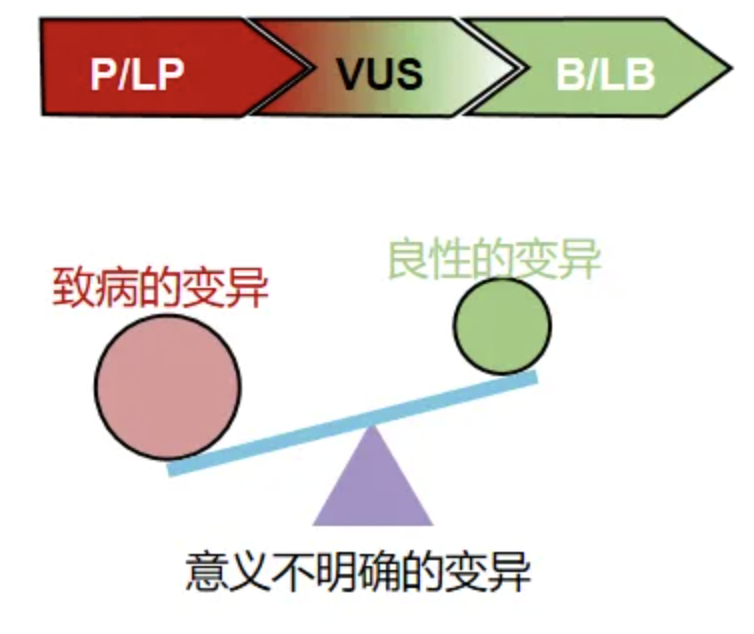
② 变异的致病性明确。ACMG(美国医学遗传学与基因组学学会)指南将变异分为5类:P(Pathogenic):致病的;LP(Likely Pathogenic):可能致病的;VUS(variant of uncertain significance):意义不明确的;LB(Likely Begin):可能良性的;B(Begin):良性的。变异的分类评估主要依据变异的类型,软件预测变异的致病性,变异在正常人群数据中的出现频率和既往报道病人情况等[9-11]。
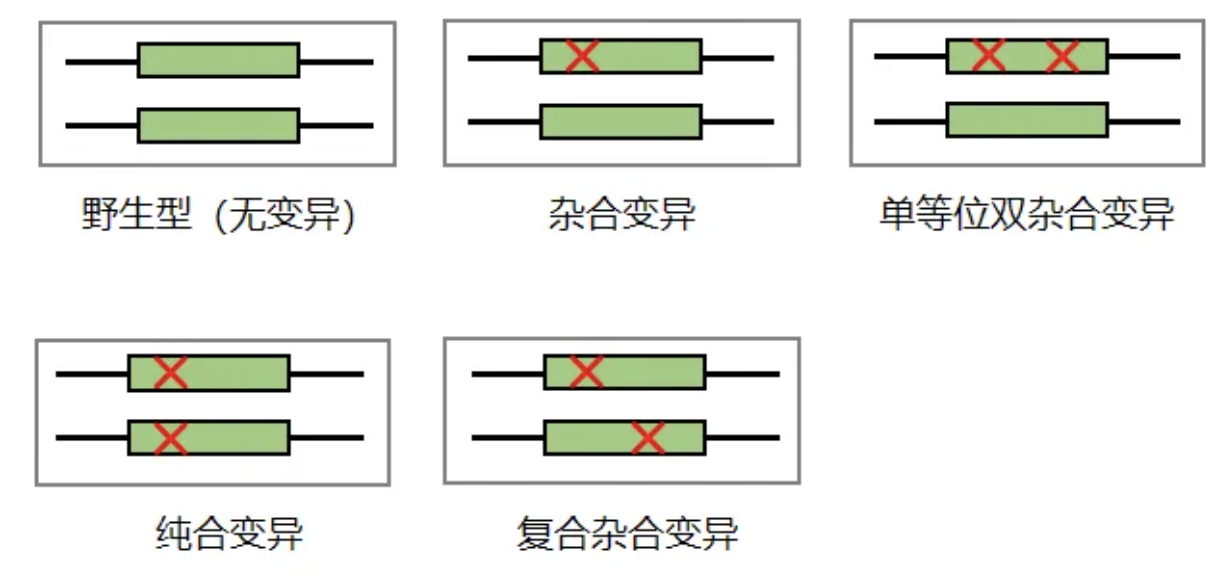
③双等位变异。人有2个等位基因(一个来自母亲,一个来自父亲)。只有2个等位基因均携带致病基因的致病性变异才会导致疾病(常染色体隐性遗传)。注:双重杂合变异,区分单等位还是双等位(复合杂合)只能通过父母或者子女进行验证,若2个变异分别来自父亲和母亲则为双等位,若2个变异均来自其中一方则为单等位。
三、基于基因变异的流行病学研究,以2个病例展示基因检测在PCD诊断的重要性
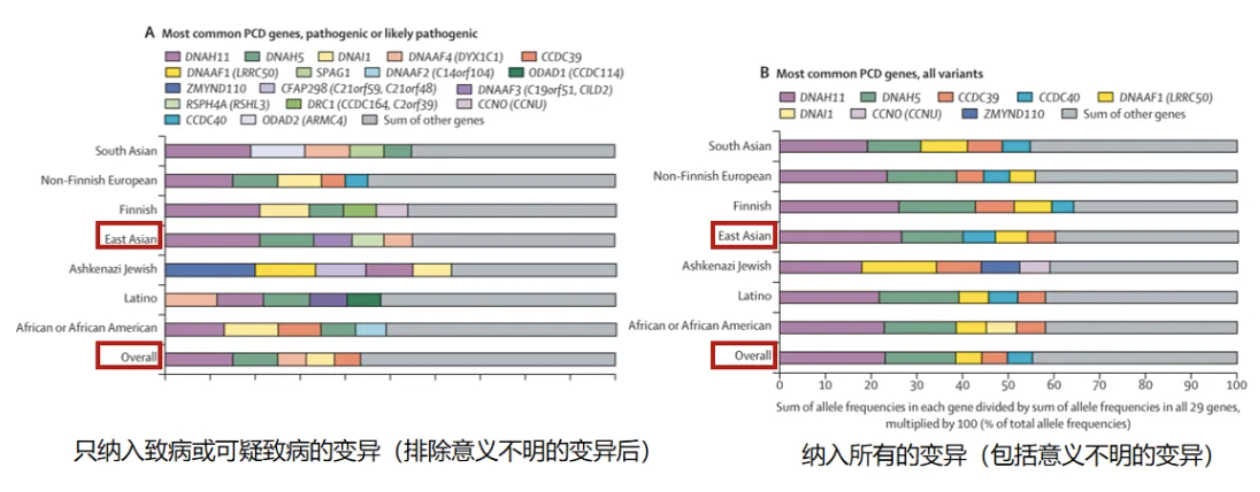
从基因型的分布可见:不同区域基因分布不同,大多数区域以DNAH11,DNAH5为主要致病基因[12]。

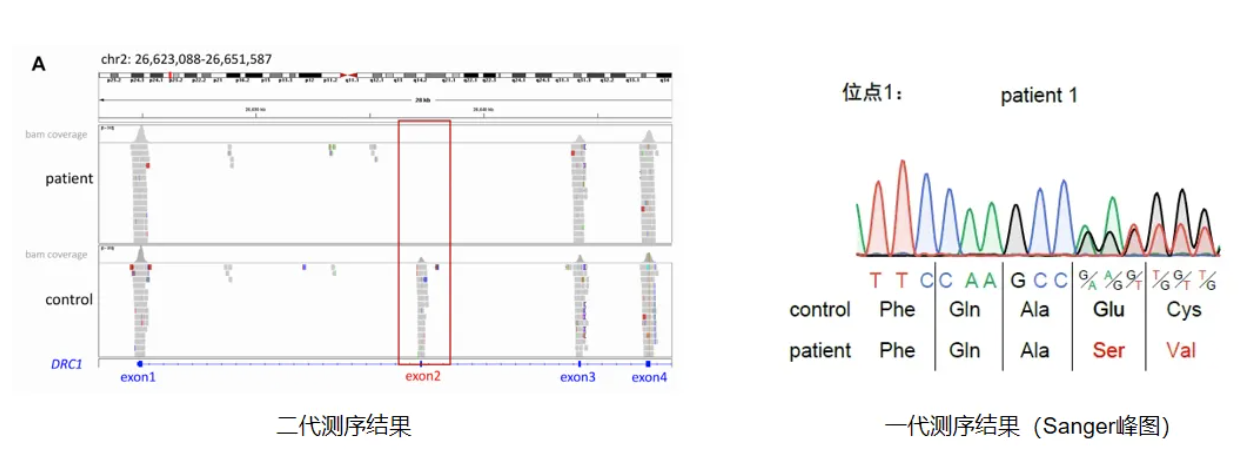
以下为我中心诊断PCD患者的两个案例。Case 1:男性患者,20岁,咳嗽咳脓痰伴鼻塞流脓涕10余年,CT显示鼻窦炎、支扩,无内脏反位。父母非近亲,妹妹体健。经鼻一氧化氮:9.0 nL/min(正常值>77 nL/min)高速视频显微镜分析:纤毛摆动极度僵硬。建议:完善父母的基因检测,检测这两个变异位点是否为双等位(Sanger测序)[13]。
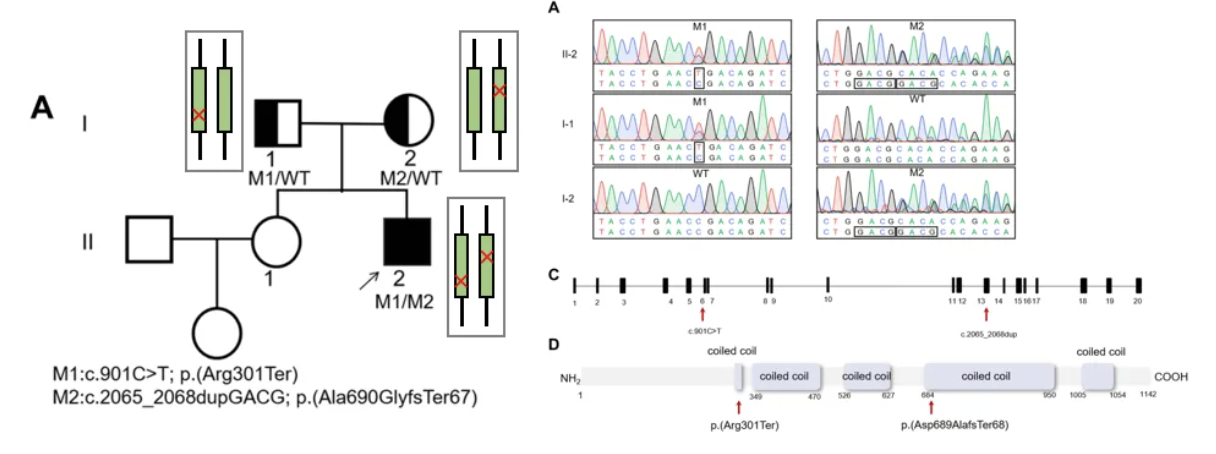
结果显示:一个变异来自父亲,一个来自母亲,符合双等位变异,确诊PCD。通过基因检测,发现患者属于CCDC40变异导致的PCD,会有弱畸精子症,属于呼吸道症状较重的类型,目前仍在继续随访中[14]。
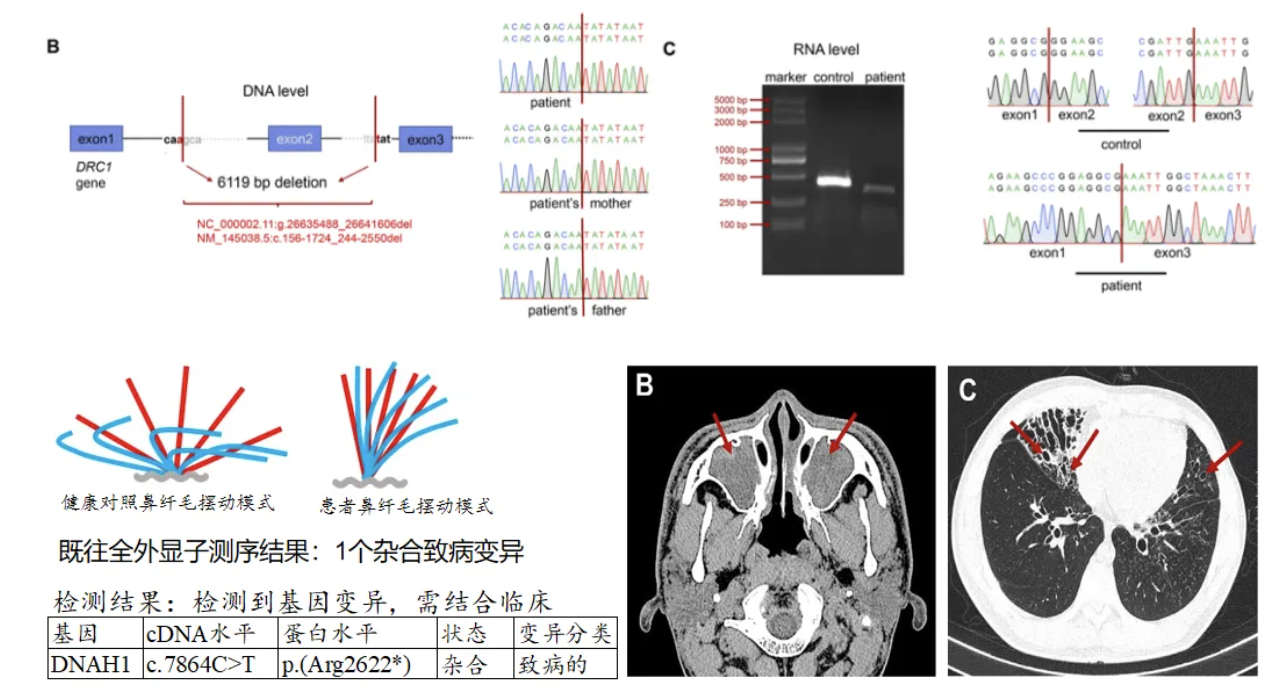
Case 2:男性患者,19岁,反复咳嗽咳痰伴鼻塞19年,有嗅觉障碍,CT显示鼻窦炎、支扩,无内脏反位。父母非近亲,姐姐体健。经鼻一氧化氮:10.8 nL/min(正常值>77 nL/min)。高速视频显微镜分析:纤毛摆动轻度僵硬。建议:目前还未确定DNAH1和PCD的因果关联,且仅检测出一个变异,无法解释患病的病情,因为患者临床症状与PCD非常相似,建议重新分析全外数据[6]。
通过基因检测,结果发现:重新分析全外显子测序原始数据,重点关注拷贝数变异,发现DRC1基因的一个大片段纯合缺失,并通过基因组DNA和鼻纤毛RNA逆转录得到的cDNA进行Sanger测序验证,最终确诊为DRC1基因变异导致的PCD[6]。
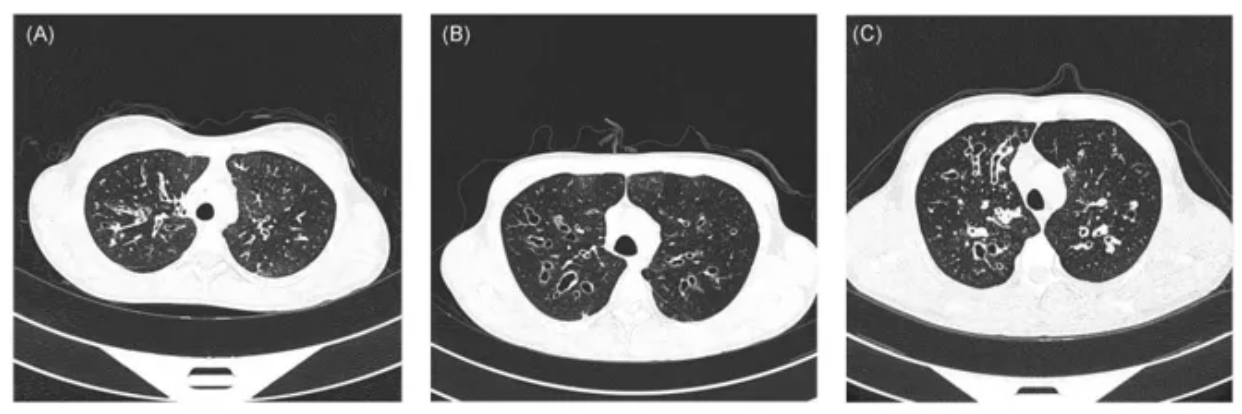
基因检测在PCD诊断的重要性:在临床上,发现高度怀疑PCD的患者,基因检测是目前普遍运用的诊断手段。对于有家族史的患者或近亲家系患者,基因检测是必要的检测手段。基因检测的优势:几乎没有操作误差;检测结果对患者及家属生育后代具有一定指导作用;有助于鉴别诊断其他可能的遗传性疾病。囊性纤维化患者常有相似的临床表现,通过基因检测可以快速确定诊断[15]。
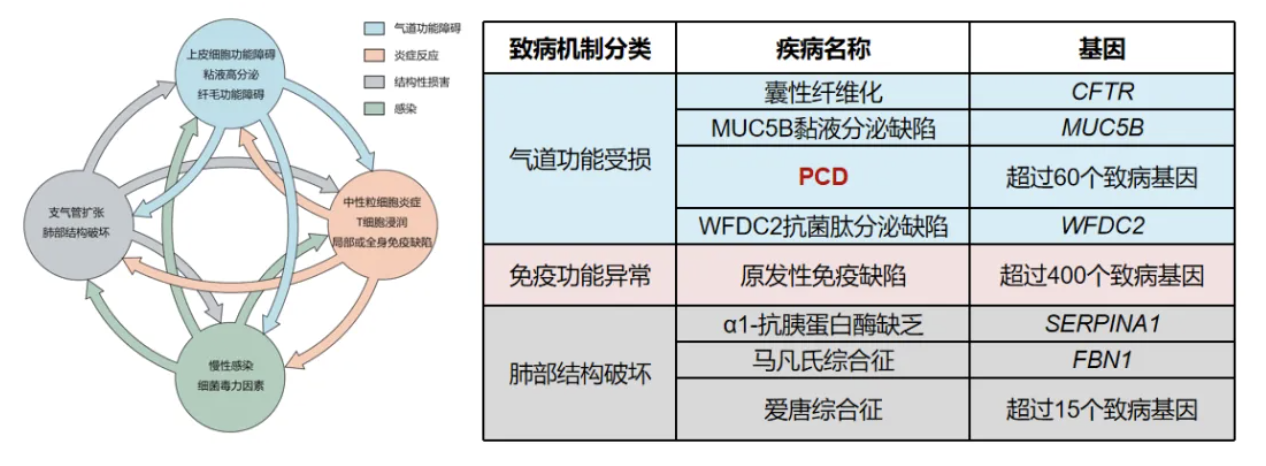
这是鉴别诊断遗传性支气管扩张症分类。对于PCD有超过60个致病基因[16]。
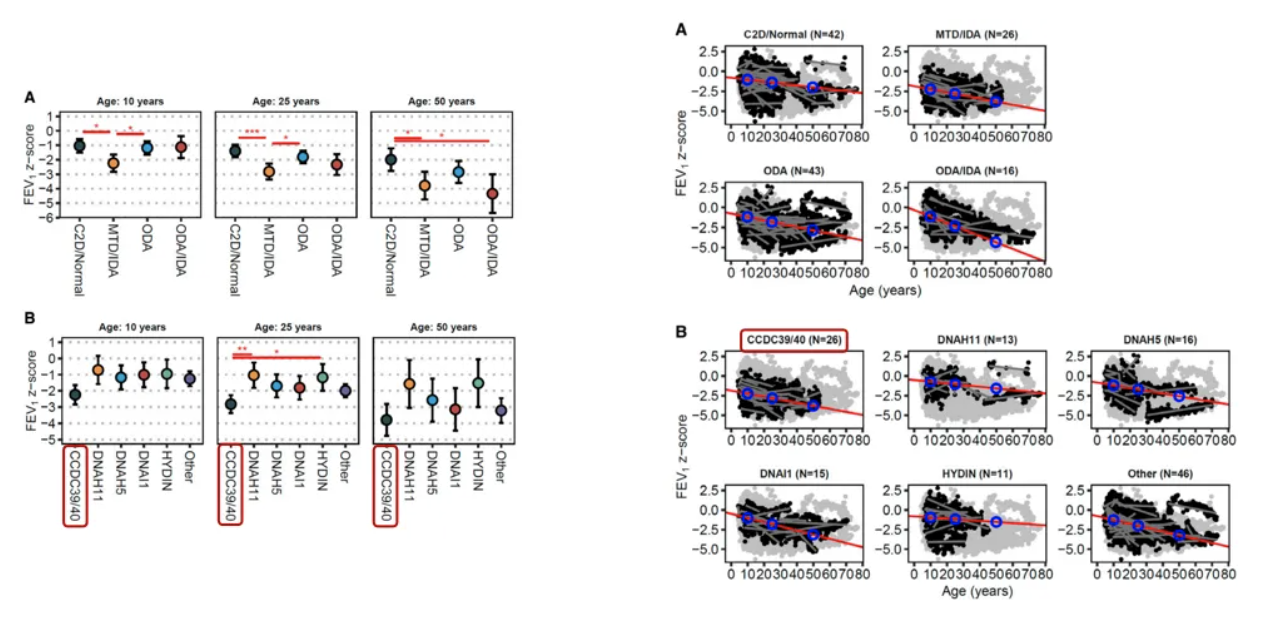
一项德国、丹麦随访43年的PCD患者队列研究比较不同结构缺陷与基因突变患者的远期肺功能改变,发现CCDC39和CCDC40突变患者更易早期出现肺功能损害,且加重程度更明显。证明基因检测结果可能提示患者疾病恶化程度[17]。
四、与纤毛相关的检查

纤毛透射电子显微镜,这项检查建议使用细胞刷采样,取材:鼻粘膜或者支气管粘膜。(强烈建议在疾病的缓解期进行采样检查)。其优点是无创、病人痛苦更小,更易接受,采样失败后方便重复采样;缺点是鼻炎可能会影响采样成功率和检测结果。而在支气管镜引导下,可选黏液较少的支气管粘膜采样,成功率更高是它的优点,但缺点是需要做支气管镜,成本较高,患者不容易接受。
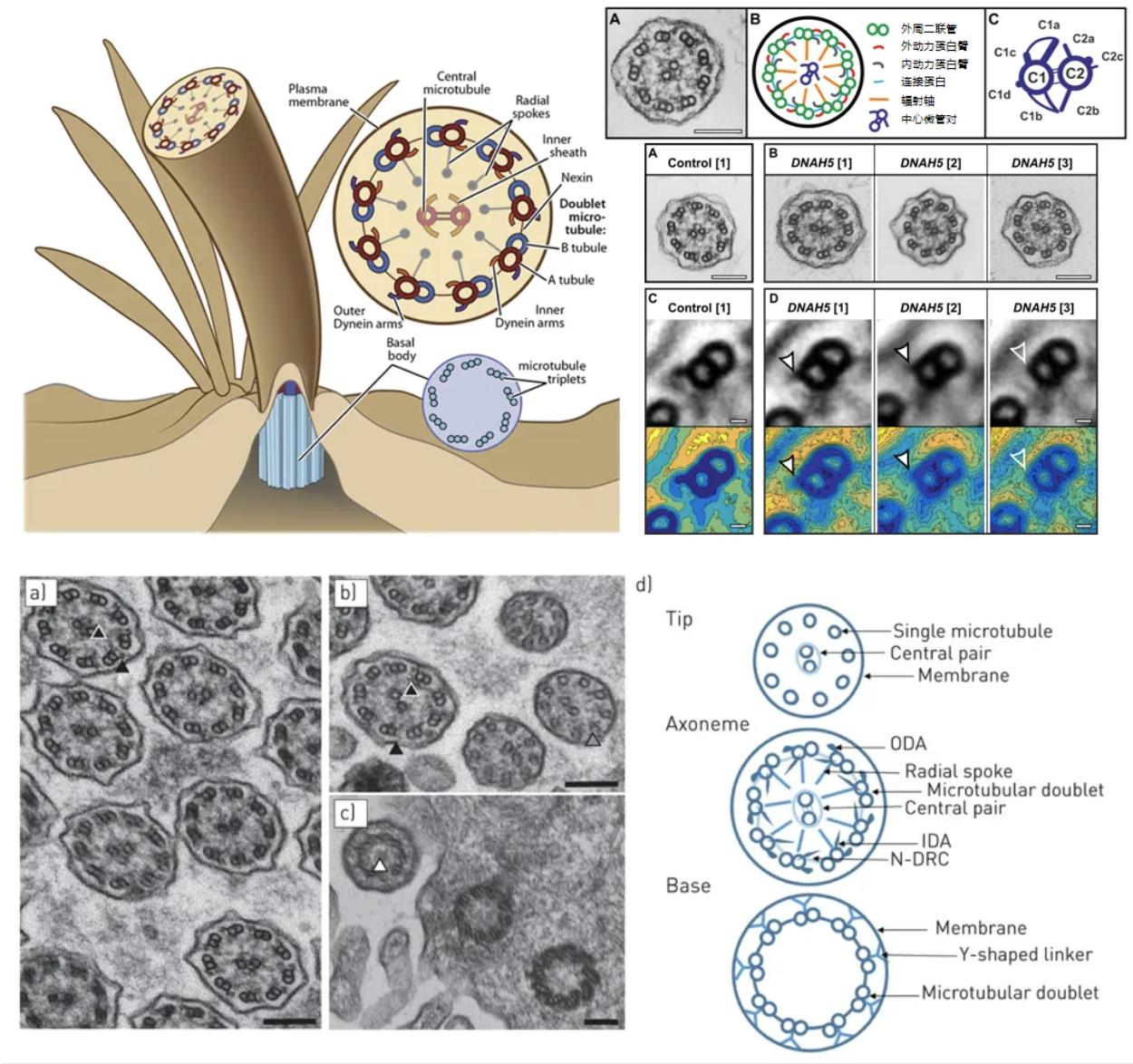
纤毛透射电子显微镜,其结果判定目前主要参考 2020 ERJ:BEAT PCD TEM Criteria。图为正常的纤毛电镜结构,在判读时需注意将纤毛尖端和基底部与异常结果进行区分[18-20]。
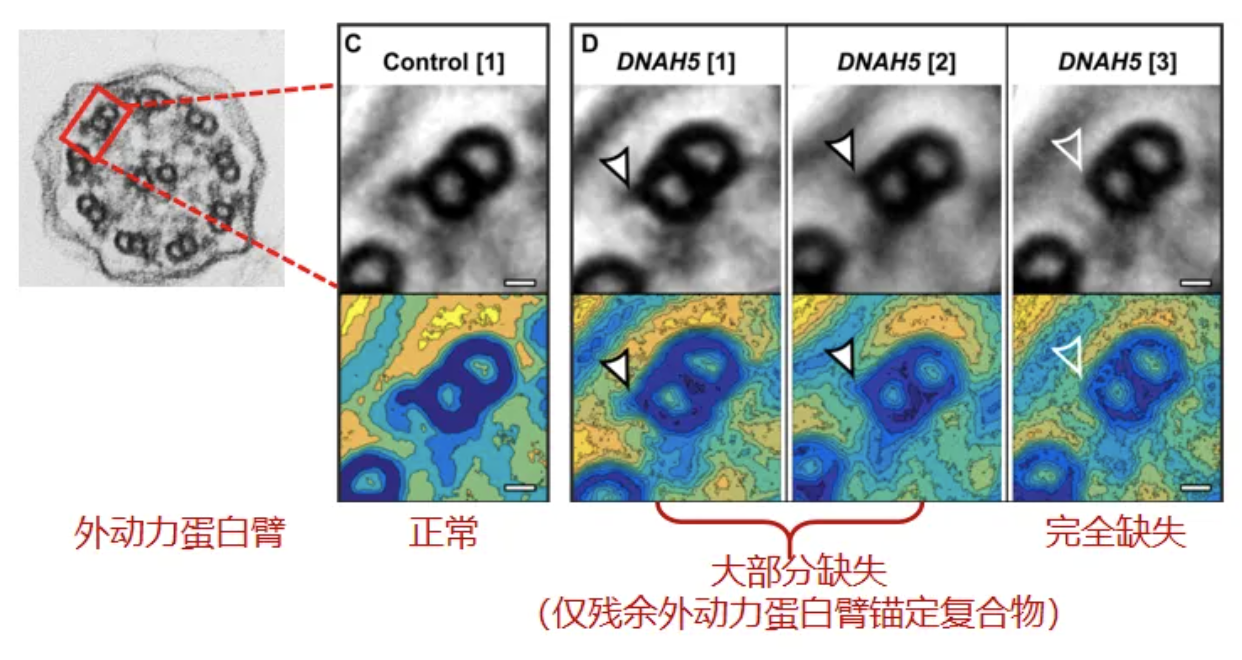
ODA(外动力蛋白臂)缺失,电镜结果评估注意:至少需要评估50个轴丝截面(多个细胞)进行,且轴丝必须有清晰的结构和完整的被膜。一个轴丝中外动力臂异常的定义:9个外周微管对中至少5个有完全缺失或部分缺失。外动力臂缺失:超过50%的轴丝有外动力蛋白臂异常[20]。
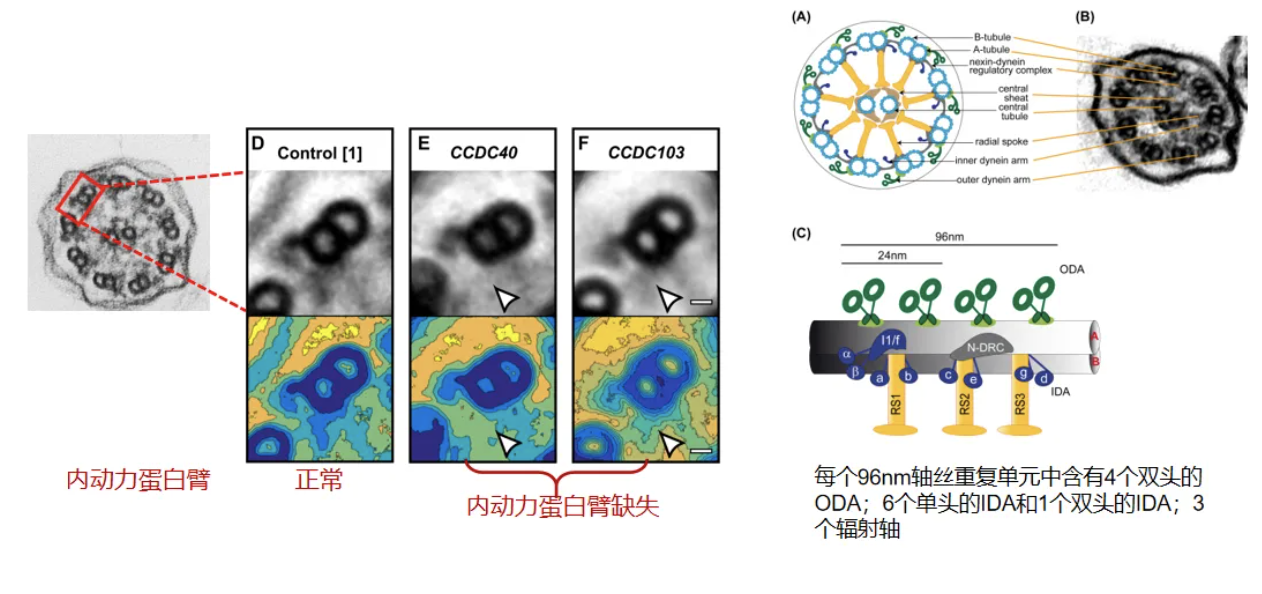
IDA(内动力蛋白臂)缺失,一个轴丝中内动力臂异常的定义:9个外周微管对中至少7个有完全缺失。内动力臂缺失:超过50%的轴丝有内动力蛋白臂异常[20]。
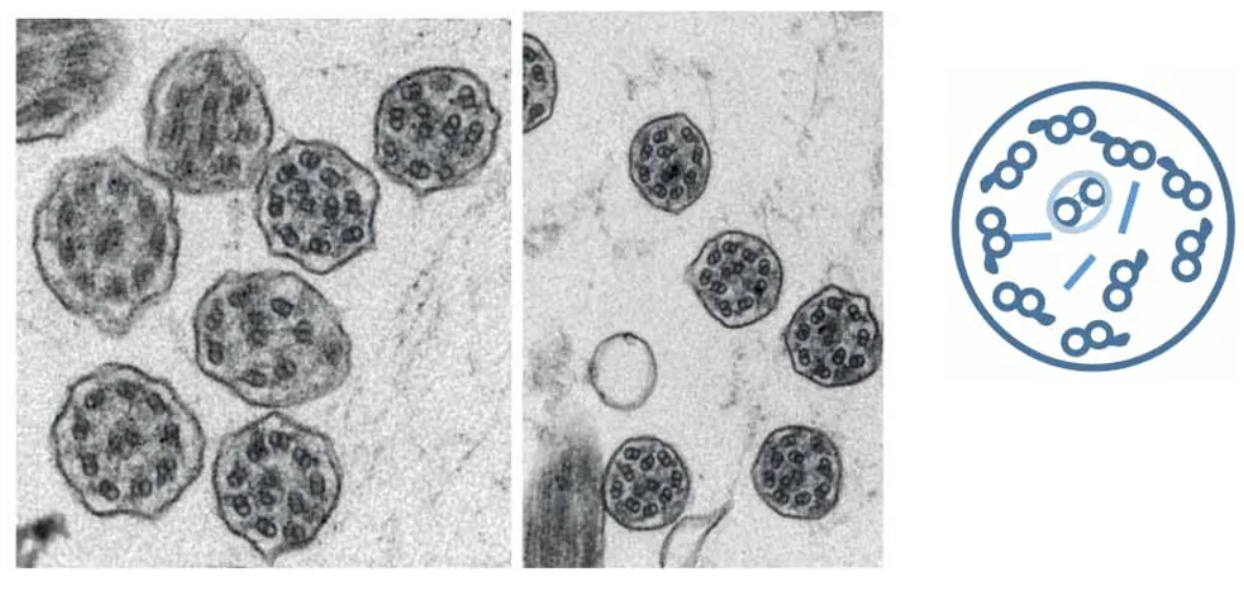
微管排列紊乱:超过25%的轴丝有9+2微管结构紊乱[20]。
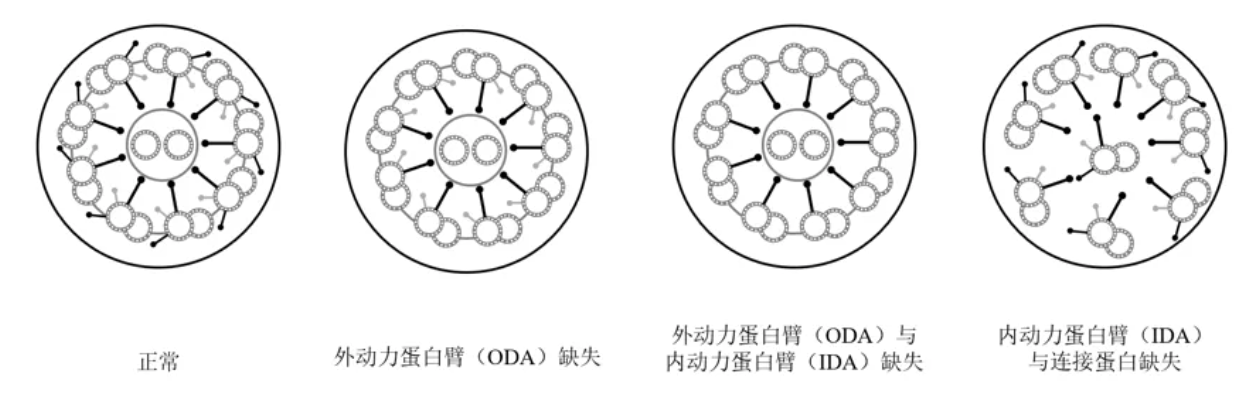
PCD异常电镜结果分类,Class1 可直接确诊PCD:外动力臂缺失、内动力臂+外动力臂缺失、内动力臂缺失+微管排列紊乱。外动力臂缺失:外动力蛋白臂编码基因变异如DNAH5,外动力蛋白臂锚定复合物基因变异如CCDC151(ODAD3);内动力臂+外动力臂缺失:动力蛋白组装相关基因如DNAAF1、DNAAF2等;内动力臂缺失+微管排列紊乱:CCDC39或CCDC40变异[20]。
关于PCD异常电镜结果分类,Class2 需要多份样本进行确认,可提示PCD,但不能确诊:中心微管对异常(超过20%轴丝):RSPH4A、RSPH1、RSPH9、DNAJB13变异可出现该异常;很少或无纤毛轴丝+基底体移位至细胞质:CCNO、MCIDAS变异可出现该异常;单纯微管排列紊乱:CCDC65、DRC1、GAS8变异可出现该异常;外动力臂缺失的轴丝比例为25%至50%:DNAH9变异(近端可正常,仅远端外动力蛋白臂缺失)可出现该异常;内动力臂+外动力臂缺失比例为25%至50%:CCDC103的错义变异p.His154Pro可出现该异常。其他异常包括纤毛肿胀、纤毛融合等与PCD关系不大,因此在PCD相关的电镜报告中可报可不报。(注:电镜结果无异常不能排除PCD的诊断)[20]。
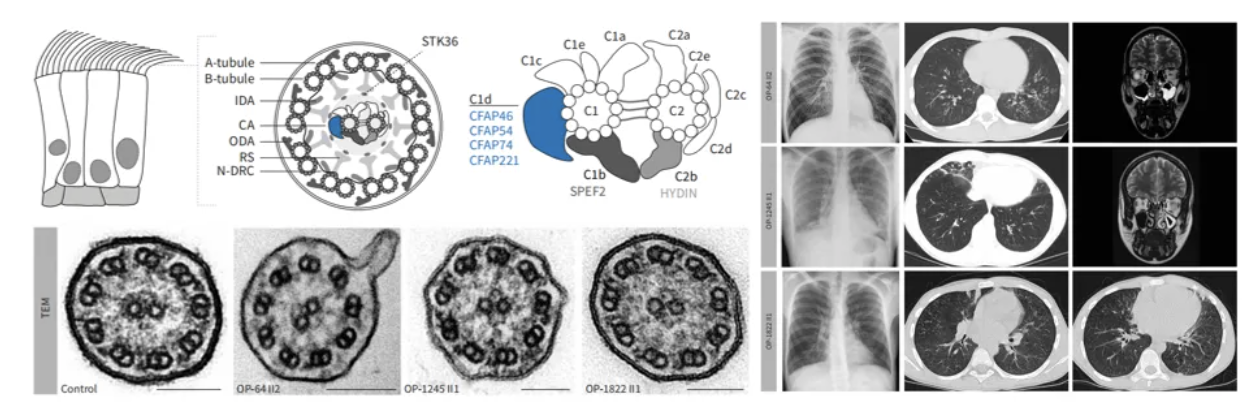
电镜结果无异常的PCD举例:CFAP46、CFAP54、CFAP74、CFAP221编码蛋白组成中央微管对C1的附属突起结构C1d,此类基因突变患者具有PCD的临床表现,但电镜超微结果未发现异常[21]。
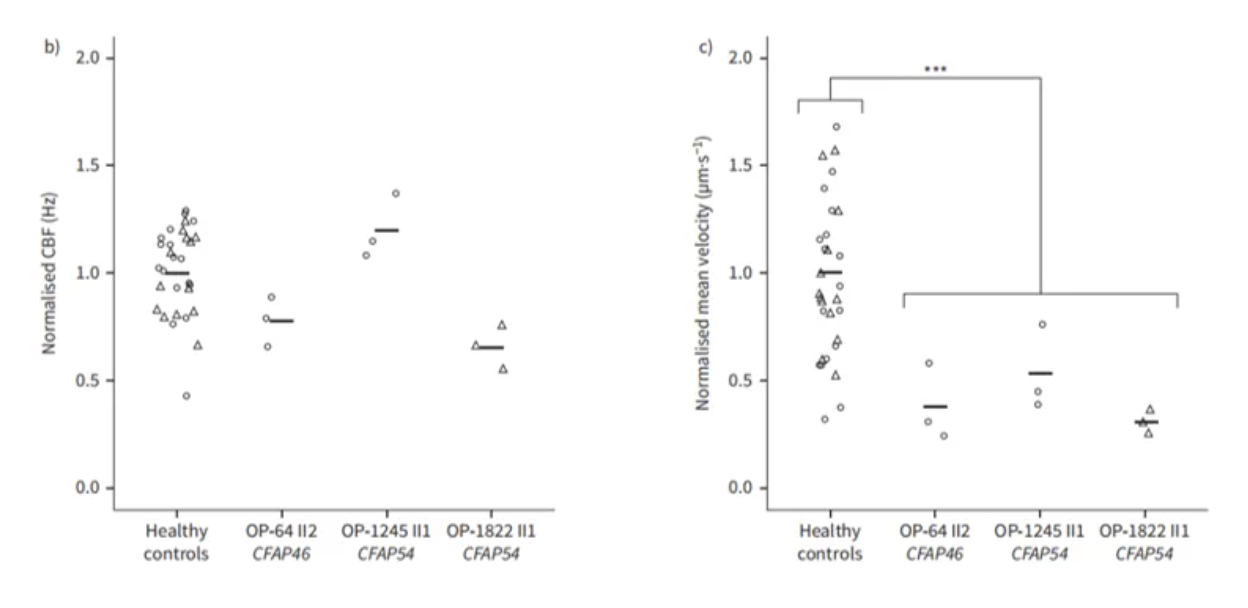
同时此类患者的HSVA分析纤毛摆动频率无明显异常,但黏液清除速率均有显著下降。对于此类电镜表现正常的PCD患者的诊断,应更加关注临床表现与基因检测结果[21]。
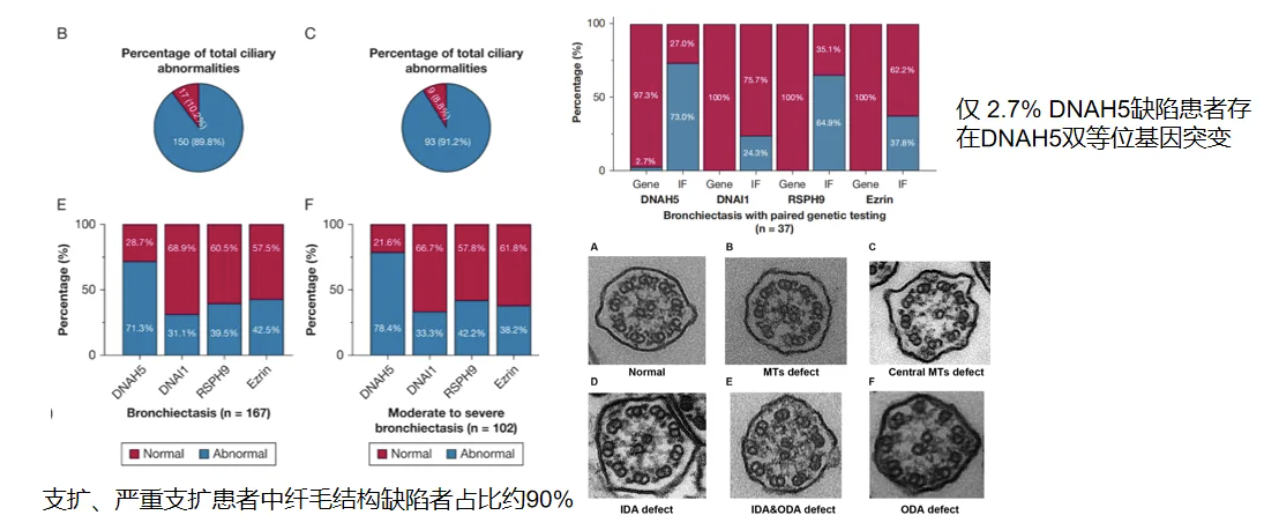
关于与继发性运动性纤毛障碍的鉴别诊断,广医关伟杰教授团队一项观察性研究结果显示支气管扩张患者大多伴随有运动性纤毛障碍(Motile ciliary disorder, MCD),TEM及IF均提示存在纤毛结构缺陷,但仅少数患者检测出基因缺陷[22]。
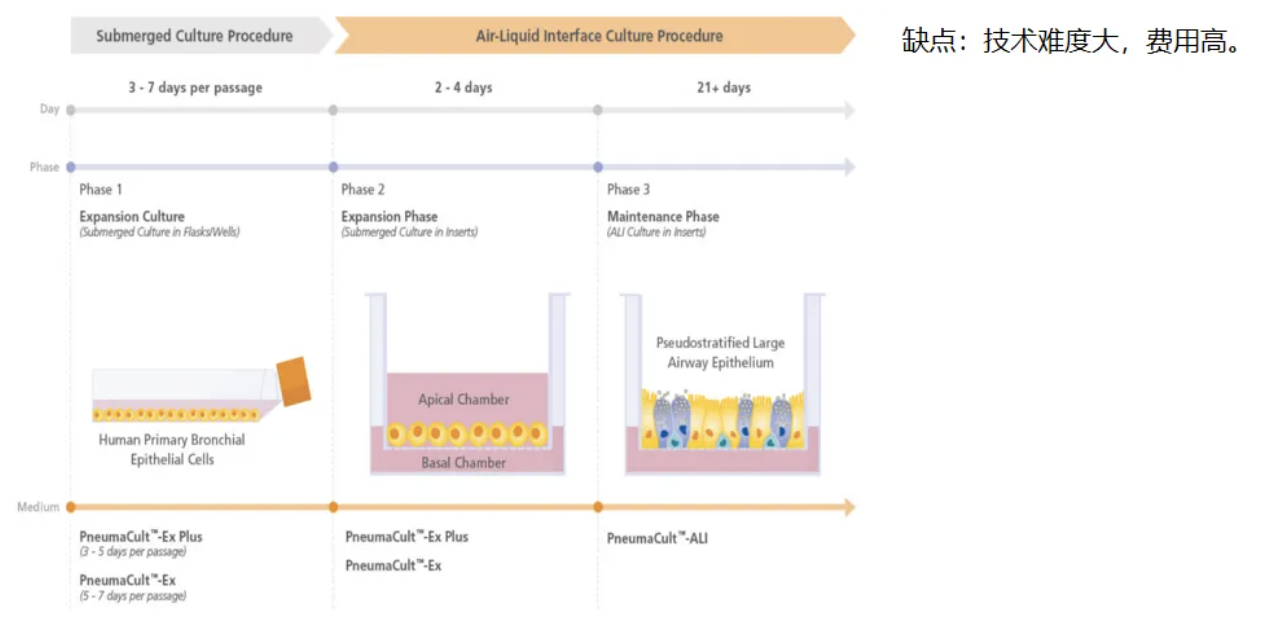
气液交界(Air-Liquid Interface, ALI)培养,通过将患者鼻粘膜或者支气管粘膜的细胞进行ALI培养,在实验室环境下进行,排除空气污染、慢性感染和炎症性损伤等继发性因素影响,可以排除部分继发性MCD,经ALI培养的细胞同样可以进行HSVA、TEM、IF等检查。
五、原发性纤毛运动障碍的预后和治疗
PCD是否会影响预期寿命?我们未发表的研究数据显示,70个明确诊断为PCD的患者中,最长随访时间为6年,其中5名患者因为肺部感染加重,呼吸衰竭而死亡。死亡年龄分别为29岁(DNAH11)、31岁(OFD1)、35岁(RSPH4A)、 47岁(DNAAF4)、30岁(CCDC40)。目前随访患者中,最大年龄患者为63岁(ODAD1基因变异患者),双肺弥漫性支扩,黏液型铜绿假单胞菌感染,每年均住院2次。而文献中的数据显示:随访的151个患者中(中位年龄为35岁),7个患者在随访的7年内死亡,死亡中位年龄为65岁(31-75岁)。因此,患者临床表现异质性非常大,疾病严重程度差异较大,部分患者预后较差。早期诊断,健康教育,日常的气道廓清治疗,积极控制感染可能是提高患者预后的关键[23]。
对于PCD治疗当前还是以对症治疗为主,对症:支气管扩张——体位引流祛痰排痰,合理运用抗生素控制感染(铜绿);不孕不育——辅助生殖手段(ICSI)。近年来相关靶向治疗实验正在进行中,距离临床实际应用仍然存在一定距离。对于早期确诊的患者,建议进行长期随访干预,预防感染,长程管理。
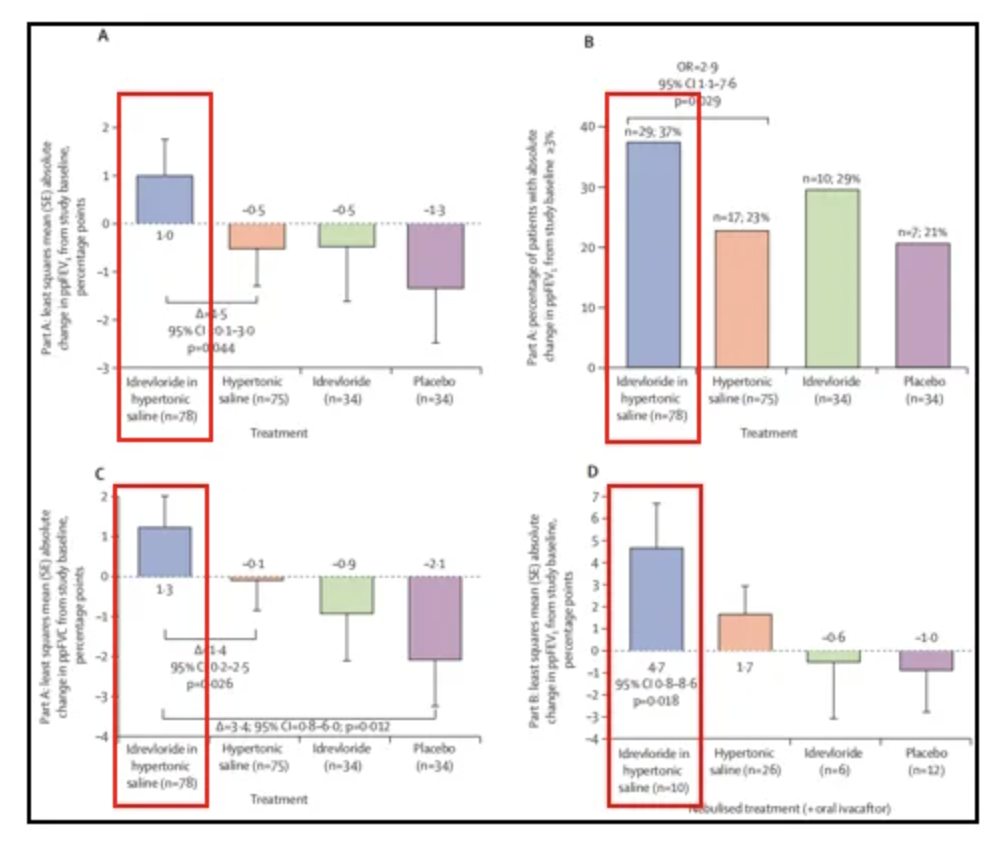
PCD治疗上的老药新用,Idrevloride:上皮钠通道阻滞剂,拮抗钠离子内流,提高胞外渗透压。高渗盐水与上皮钠通道阻滞剂idrevloride合用较单独使用高渗盐水在28天内对PCD患者的肺功能改善作用更加明显,且不会增加安全风险[24]。

关于靶向治疗的探索,有DNAI1细胞试验,显示经过优化的mRNA脂质纳米粒(雾化)的蛋白表达水平更高,细胞炎症反应更低。DNAI1 mRNA脂质纳米粒(雾化)能在体外细胞模型中恢复纤毛的摆动,效果可持续数周[25]。
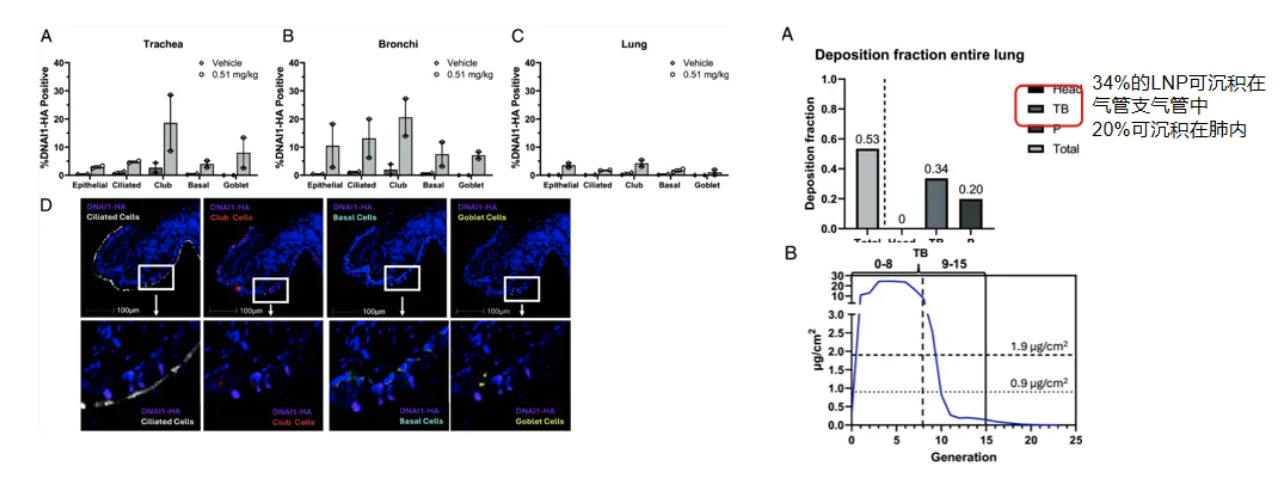
这是DNAI1动物试验。经插管向食蟹猴气道内吸入输送DNAI1 mRNA脂质纳米粒可以在食蟹猴气管、支气管与肺内检测到DNAI1表达,提示该疗法具备成功的可能性。其局限性是未构建PCD疾病模型评估治疗情况[25]。
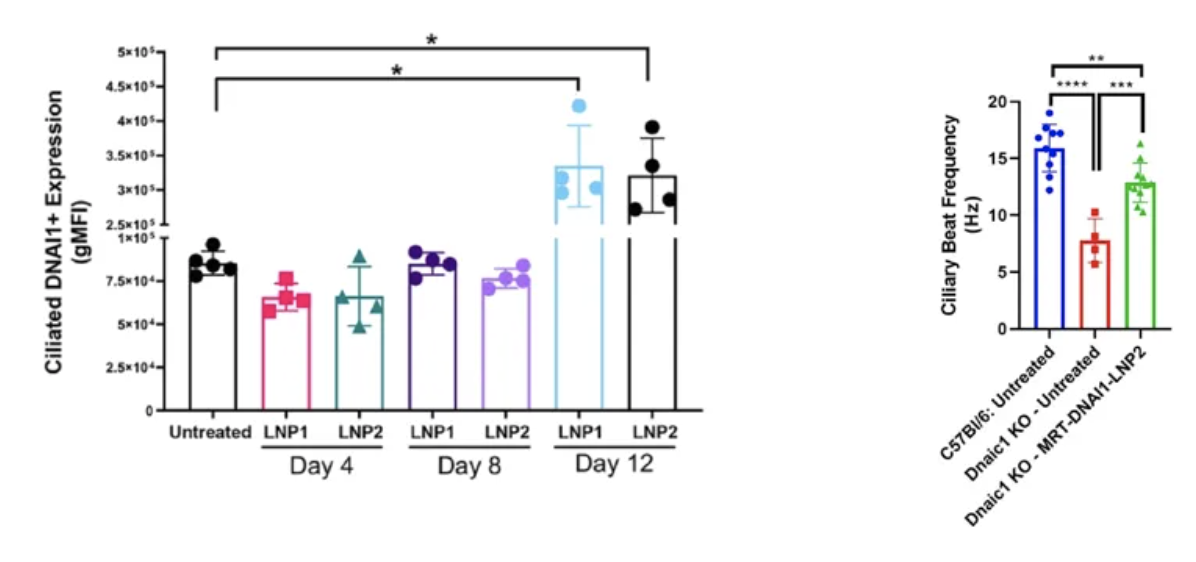
通过雾化吸入的方法,将编码DNAI1的优化mRNA包裹在脂质纳米粒中,并在纤毛细胞中表达。局限性:PCD小鼠模型不具备完整的肺部疾病表现[26]。
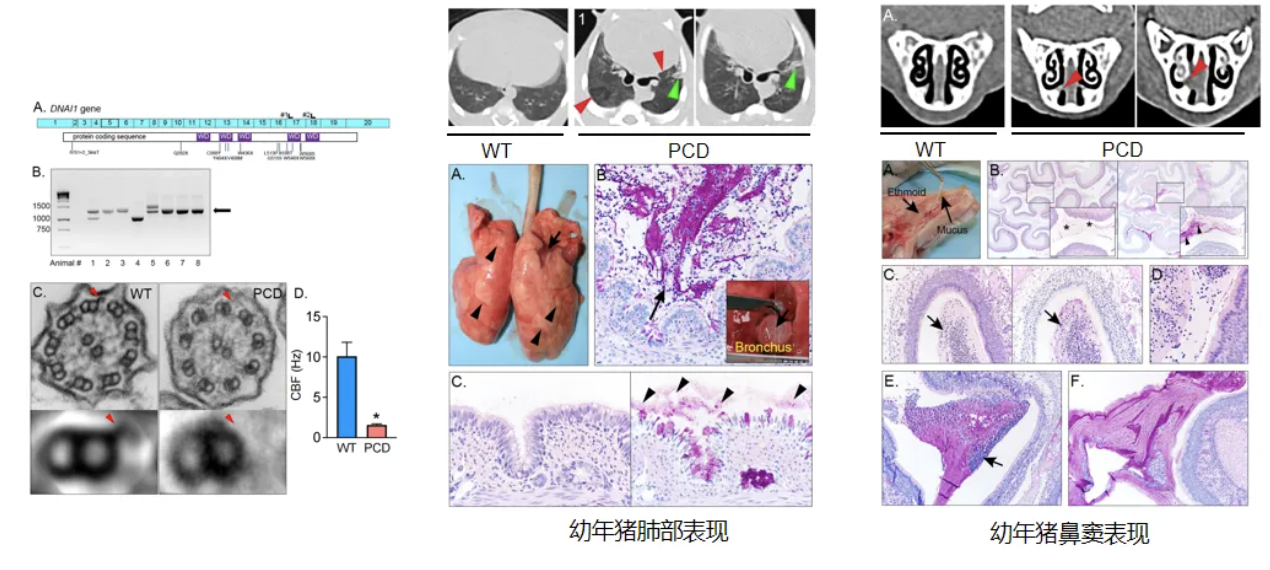
这是PCD猪动物模型,美国爱荷华大学运用CRISPR-Cas9技术构建DNAI1基因敲除猪,幼年时观察到肺叶塌陷不张、气道与鼻窦黏液聚积,但无明显感染[27]。
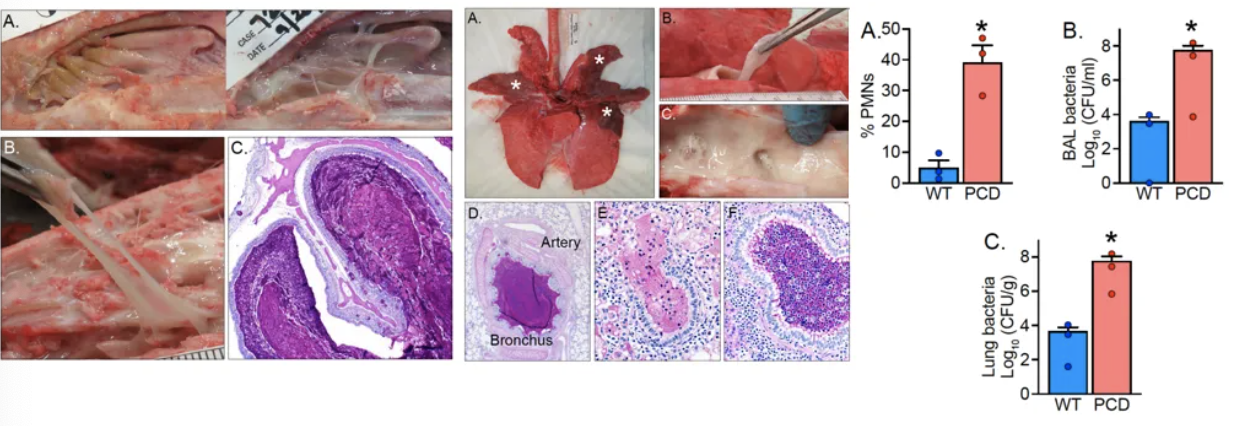
年长的DNAI1敲除猪可观察到明显的肺部炎症表现与黏液聚积。猪气道解剖和生理学与人类更相似,PCD猪动物模型可以出现明显的上下气道疾病表型,为PCD机制研究与靶向治疗探索提供一个新途径[27]。
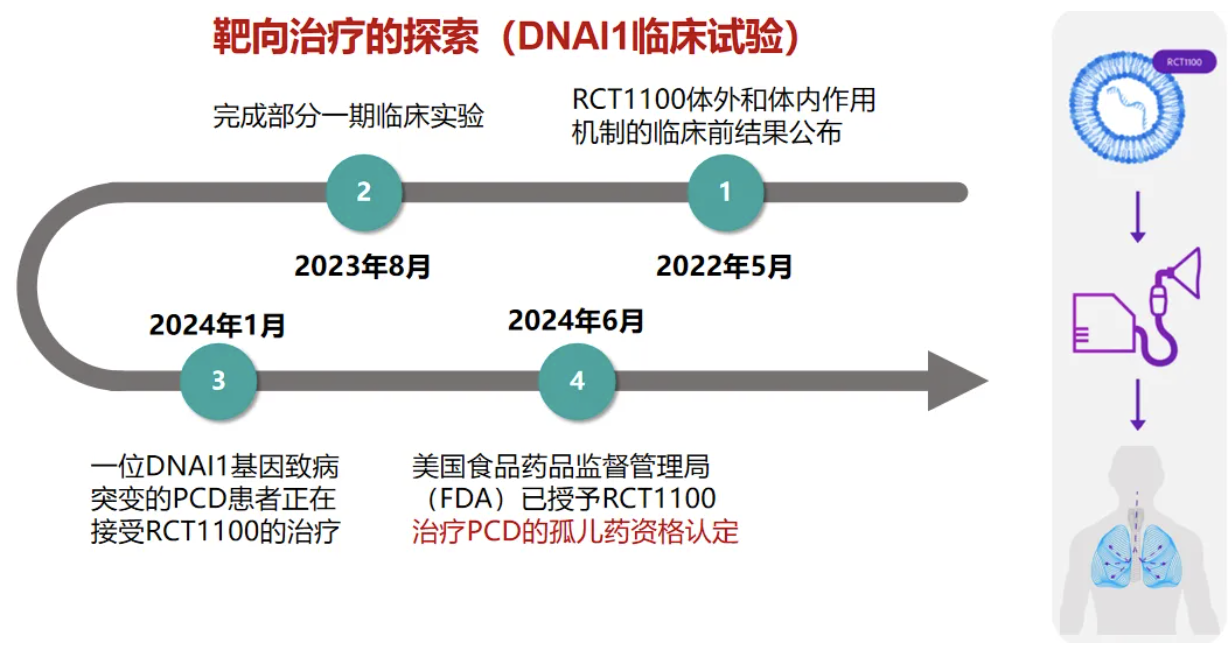
这是DNAI1临床实验的整个历程。在2024年6月,美国食品药品监督管理局(FDA)已授予DNAI1靶向治疗药物RCT1100治疗PCD的孤儿药资格认定。
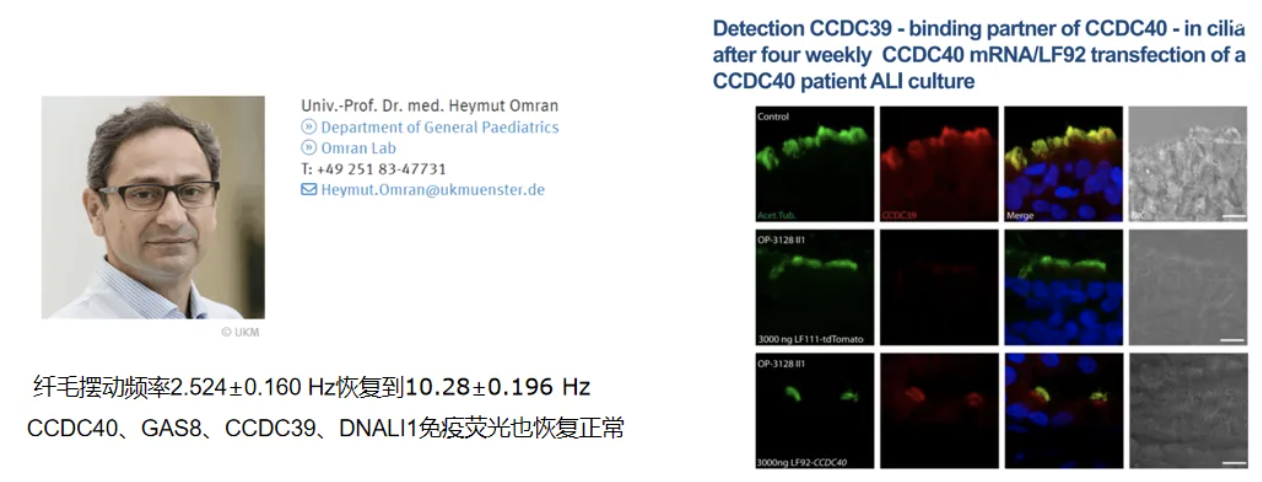
CCDC40也是靶向治疗的探索。Ethris公司在mRNA疗法治疗肺部疾病的方面的领先地位,具有非免疫原性修饰mRNA 和类脂质纳米颗粒 (LNP) 递送平台。CCDC40患者的离体鼻纤毛细胞mRNA治疗6周,成功恢复了纤毛的结构和功能[28-29]。
综合以上研究进展可以看出,基因治疗必然是未来的方向。
总结
1.PCD是我国遗传性支扩中最常见的病因,以呼吸道表现为主,常伴随有多器官系统表现;
2.鼻呼气一氧化氮(nNO),高速视频显微镜(HSVA)和免疫荧光是有效的PCD检查手段;
3.基因检测和电镜检测结果可确诊PCD,但需要关注结果的解读,错误的解读可能会导致PCD的误诊;
4.基因检测是当前我国诊断PCD的主要方法,我国PCD患者最常见的基因型为DNAH11和DNAH5;
5.目前对原发性纤毛运动障碍治疗方案以气道廓清与控制感染为主,靶向治疗方法尚在探索阶段,预后不佳。
参考文献
1. 上海医学, 2020,43(04):193-202.
2. 中华实用儿科临床杂志, 2018, 33(2), 94-99
3. Am J Respir Crit Care Med. 2018;197(12):e24-e39.
4. Eur Respir J. 2017;49(1):1601090.
5. Am J Respir Crit Care Med. 2017 Jul 1;196(1):94-101.
6. Front Genet. 2022;13:940292.
7. Hum Mutat 2012; 33: 495-503.
8. Chest 2021; 159: 1768-81
9. Circ Cardiovasc Genet. 2017;10:e001839
10. Genet Med 2015; 17: 405-24.
11. 中国科学:生命科学 2017; 47: 668-88.
12. Lancet Respiratory Medicine 2022; 10:459-68.
13. Pharmgenomics Pers Med 2022; 15: 341-50.
14. Pharmgenomics Pers Med 2022; 15: 341-50.
15. Front Genet. 2021 Jul 2;12:631221
16. Lancet, 2018,392(10150):880-890.
17. Ann Am Thorac Soc. 2025 Feb;22(2):216-225.
18. https://doi.org/10.1007/978-3-030-59265-3_30
19. Am J Physiol Lung Cell Mol Physiol319: L1048–L1060, 2020.
20. Eur Respir J. 2020, 55(4):1900725.
21. Eur Respir J. 2024 Dec 12;64(6):2400790.
22. Chest vol. 163,5 (2023): 1038-1050.
23. European Respiratory Journal, 2016, 48(2), 441-450
24. The Lancet. Respiratory medicine vol. 12,1 (2024): 21-33.
25. Proc Natl Acad Sci U S A. 2025 May 6;122(18):e2421915122.
26. https://doi.org/10.1016/j.pupt.2022.102134
27. bioRxiv : the preprint server for biology 2024.05.22.594822. 21 Aug. 2024
28. Klin Padiatr 2023; 235(02): 131
29. https://www.thieme-connect.de/products/ejournals/abstract/10.1055/s-0043-1761570
专家介绍

罗红
教授,一级主任医师,博士生导师;中南大学湘雅二医院呼吸与危重症医学科主任,危重症亚专科主任;中国医师协会呼吸医师分会常委;中华医学会呼吸病学分会呼吸危重症学组委员;中华医学会呼吸病学分会呼吸遗传及罕见病筹备学组副组长;湖南省医师协会呼吸医师分会会长;湖南省医学会呼吸病学专业委员会副主任委员;湖南省基层呼吸疾病防治联盟主席;主持国家自然科学基金面上项目4项;全国五一劳动奖章获得者。
本文由《呼吸界》编辑 冬雪凝 整理,感谢罗红教授的审阅修改!
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry