

 分享
分享

引言
该案例在疾病诊断上因「症状多样且不典型,缺乏特异性诊断方法、组织病理学检查」而颇具挑战,又因目前缺乏大规模的临床研究来指导治疗,因此治疗方案的选择通常基于医生的经验和患者的具体情况,可能导致治疗效果有所差异。这种疾病的病变程度和范围不同,需根据患者的具体情况进行分期和分级,且治疗上也存在一定的副作用,同时还有较高的复发率,缺乏有效的治疗药物。通过该案可告诉我们,患者的哪些生活行为、病变表现需要与哪些疾病进行鉴别诊断?后续治疗方案还应考虑哪些细节?
无诱因反复出现「干咳」和「活动后气短」,双肺多发囊腔病灶……肺朗格汉斯细胞细胞组织细胞增生症?
卷宗1
基本资料:男性患者,37岁,陕西清涧县人,主诉「干咳2月余,气短3天」于2021年10月27日入住我院呼吸与危重症医学科。
讲述者:臧宇
2021年8月下旬,患者无明显诱因出现「阵发性干咳」,咳嗽无明显昼夜规律,与吸入冷空气或其他刺激性气体无关,与体位变化或呼吸运动也无关,也无「发热、咳痰、流涕、鼻塞」,无夜间阵发性呼吸困难,无反酸、烧心、呃逆,因此当时完全未重视,没有上医院诊治。但一段时间后,患者的间断咳嗽症状毫无好转(性质基本同前),于是在8月27日到当地县医院就医。
胸部CT+肋骨三维重建:双肺多发囊腔病灶,以双上肺为主,考虑肺朗格汉斯细胞细胞组织细胞增生症可能,当地医院建议结合临床及实验室检查;胸骨柄右侧缘可见局限性透亮区,考虑累及胸骨柄。医院给予了止咳平喘类药物治疗(具体不详),患者症状好转后出院。但两个月后,也就是患者到我院就诊的3天前,又出现了无明显诱因的「活动后气短不适」,随后我院以其「双肺囊性病变」收入院治疗。
患者自起病以来,精神、睡眠一般,食欲尚可,二便正常,体重未见明显变化。查既往史,患者1年前出现过腹部皮疹,自行涂抹膏药治疗1月后好转;半年前出现过「干咳2周」,没有诊治便自行好转。患者否认慢性病、传染病史,否认手术、外伤、输血、中毒史,对「磺胺类」药物过敏;吸烟史18年,约15支/天;偶有饮酒;无放射物、药物、毒物接触或暴露史;长期从事公司职员,无不洁饮食史。家族史无特殊。入院后检查请见第二份卷宗。
卷宗2
体格检查:
T 36.3℃,P 78次/分,R 20次/分,BP 128/78mmHg,SPO2 97.1%(未吸氧)。精神可,意识清,体重75kg,身高178cm,BMI指数23.67kg/m2,发育正常,查体配合。全身皮肤无皮疹、出血点。口唇红润,口腔黏膜正常,咽部无充血,双侧扁桃体无肿大。左侧颈部可触及数个肿大淋巴结,相互融合,与周围组织分界不清,质地韧,无压痛,局部皮肤无红肿或破溃,双侧腹股沟及右侧腘窝淋巴结可触及,直径约1cm,表面光滑,质地柔软,与周围组织无粘连,无压痛,表面皮肤正常,余部位浅表淋巴结触诊无异常。胸廓无畸形,双侧呼吸动度对称,语颤无增强,双肺叩诊清音,双肺呼吸音清晰,双肺未闻及干湿性啰音,未闻及胸膜摩擦音。
心前区无隆起,心尖搏动位于第五肋间左锁骨中线内0.5cm,未触及细震颤,心界无扩大,心率78次/分,律齐,心音无明显增强和减弱,各瓣膜听诊区未闻及病理性杂音。腹部平软,无肝脾肿大,全腹无压痛及腹肌紧张。脊柱、四肢关节无畸形,无肿痛及运动障碍,肌肉无压痛及萎缩。双下肢无水肿。
实验室检查如下:
1.常规指标
【动脉血气分析】PH 7.395,pCO2 44.1mmHg,pO2 101mmHg,HCO3- 26.4mmol/L, 氧合指数480。
【血、尿、便常规+潜血试验】正常
【炎症指标】CRP、hs-CRP、PCT正常
【生化指标】
肝功八项:ALB 53.4 g/L↑,GLO 16.4 g/L ,A/G 3.3↑,余正常;
肾功、电解质、NTpro-BNP、肌钙蛋白I、肌红蛋白、CK-MB正常
【凝血功能】正常
【甲状腺功能八项】正常
2.病原学及肿瘤相关指标
【感染八项】乙肝2对半、丙肝、HIV、梅毒(-)
【肺部肿瘤标志物】正常
3.风湿免疫相关指标
【ANCA系列】:pANCA、cANCA、MPO、PR3均阴性
【抗核抗体滴度+自身抗体ANA谱】阴性
【风湿系列】抗CCP抗体、抗链球菌溶血素O、类风湿因子正常;
【总IgE】 345IU/ml ↑;
【免疫球蛋白补体系列】补体C1抑制因子0.43g/L↑,血清IgG、IgA IgM 正常,K-轻链、λ-轻链、K/λ、补体C3裂解产物、补体C4 正常
4. 常规心电图:大致正常

患者入院时的胸部增强CT
这是患者2021年10月27日的入院胸部增强CT,显示:双肺散在大小不等囊腔,边缘略模糊;双肺散在结节状、斑片状磨玻璃影;右肺上叶后段可见一混合磨玻璃结节,大小约2.3cm*2.3cm,形态不规则,边界不清晰,其内见直径约0.5cm实性结节影。同日进行的肺功能检查,显示:通气功能正常,肺弥散功能正常,支气管舒张试验阴性。FENO 24ppb。彩超、支气管镜检、病理及穿刺活检结果请见第三份卷宗。
卷宗3
彩超:
【心脏及腹部超声】心脏彩超未见异常;肝、胆、胰、脾、双肾、双侧肾上腺区未见异常,腹腔未见明确肿大淋巴结。
【全身浅表淋巴结】左侧颈部可见数个大小不等的低回声结节,边界清晰,形态规则,内可见淋巴门强回声,皮质增厚,较大的约0.8cm*2.0cm,CDFI示内见点条状血流信号;双侧腹股沟区淋巴结可见(左侧较大的0.4cm*0.9cm,右侧较大的0.4cm*1.0cm);右侧腘窝淋巴结增大,约0.8cm*0.8cm,边界清晰,形态规则,淋巴们结构消失;右侧颈部、双侧锁骨上窝、双侧腋窝、左侧腘窝未见明确肿大淋巴结。
电子支气管检查:镜下各管腔未见异常;根据胸部CT定位于右肺上叶后段处给予超声探查发现密度均匀低回声区,周围无血管伴行,予超声引导下经支气管镜肺活检3块组织送检,并行保护性毛刷刷检。(2021-10-22)
肺泡灌洗液结果:
【病原学】呼吸道13联病原菌核酸检测阴性;分枝杆菌菌种鉴定芯片结果阴性;细菌涂片查见G+杆菌、G-杆菌,查见G+球菌,未查见G-球菌;抗酸染色未见抗酸杆菌;细菌培养阴性
【细胞病理】肺泡灌洗液可见中性粒细胞、淋巴细胞,未见病原体,未查见瘤细胞(2021-10-22)
毛刷刷检病理:
右肺上叶后段刷检可见中性粒细胞、柱状细胞、淋巴细胞、组织细胞,未见病原体,未查见瘤细胞(2021-10-26)
支气管镜活检病理:
右肺上叶后段支气管粘膜-肺组织慢性炎伴炎性渗出及纤维组织增生
超声引导下右侧腘窝淋巴结穿刺活检:
送检少许纤维脂肪组织、血管、横纹肌及滑膜组织,小组织穿刺标本,有一定局限性,建议结合临床。
回顾该患者的症状和体征:间断干咳2月余,活动后气短3天,无明显加重或缓解因素,无胸痛、咳痰、咯血,无发热、盗汗、消瘦、全身乏力等不适,症状无特异性。患者口唇红润,全身无皮疹、出血点,左侧颈部可触及数个肿大淋巴结,相互融合,与周围组织分界不清,质地柔软,无压痛,局部皮肤无红肿或破溃,双侧腹股沟及右侧腘窝淋巴结可触及,直径约1cm,表面光滑,质地柔软,与周围组织无粘连,无压痛,表面皮肤正常,余部位浅表淋巴结触诊无异常。胸廓无畸形,双侧呼吸动度对称,语颤无增强,双肺叩诊清音,双肺呼吸音清晰,双肺未闻及干湿性啰音,未闻及胸膜摩擦音。心脏及腹部查体无异常。辅助检查详见第四份卷宗:
卷宗4
辅助检查:
①胸部CT:双肺散在大小不等囊腔;双肺散在结节状、斑片状磨玻璃影;右肺上叶后段可见一混合磨玻璃结节,大小约2.3cm*2.3cm,形态不规则,边界不清晰,其内见直径约0.5cm实性结节影。
②入院血气分析、三大常规、生化指标、炎症感染指标、甲功、自身免疫相关指标均未见明显异常。
③肺功能、FeNO检测正常
④电子支气管镜下肺泡灌洗液及支气管镜保护性毛刷刷检均提示病原学检测阴性,未查见瘤细胞。
⑤电子支气管镜下观未见异常,EBUS-TBNB结果示右肺上叶后段支气管粘膜-肺组织慢性炎,伴炎性渗出及纤维组织增生。
⑥右侧腘窝淋巴结活检未见异常。
总结该患者的病例特点,青年男性,慢性病程,间断干咳2月余,活动后气短3天入院,胸部CT提示双肺散在多发大小不等囊性病灶,主要分布于双上肺;双肺散在结节状、斑片状磨玻璃影;右肺上叶后段可见一直径约2.3cm的混合磨玻璃结节。本例以影像表现为肺囊性病变为切入点进行分析,诊断思路如下:

肺囊性病变诊断思路
对该患者需要进行10种疾病的鉴别诊断,首先是过敏性肺炎,支持点:患者有干咳、气短,症状无明显加重或缓解,HRCT显示多发磨玻璃影及多发肺囊肿灶;不支持点:无干草、鸟禽类等暴露病史, BALF淋巴细胞比例正常。
脱屑性间质性肺病(DPI)?支持点:患者慢性起病,临床表现为干咳、活动后气短,既往长期吸烟史,HRCT双肺散在囊腔及磨玻璃影;不支持点:HRCT显示病灶多分布于双肺中上部,而非双肺基底部和胸膜下。
会是呼吸性细支气管炎-间质性肺病(RB-ILD)?支持点:长期吸烟史,亚急性病程,主要症状为干咳,逐渐出现活动后气短,HRCT显示多发散在磨玻璃影、囊腔;不支持点:肺部听诊正常,肺活检可发现呼吸性细支气管内有色素沉着的巨噬细胞。间质性肺炎还有一种叫淋巴细胞性间质性肺炎,会是这种疾病吗?支持点:HRCT可见多发磨玻璃影,伴薄壁囊腔形成;不支持点:患者无自身免疫性疾病史,免疫相关指标无异常,薄壁囊腔直径相对较小且多分布于双肺中上部。
卡氏肺孢子菌肺炎(PCP)?支持点:患者以干咳起病,伴活动后气短,肺部查体无异常,浅表淋巴结肿大,双肺散在多发磨玻璃影;不支持点:患者无免疫抑制的诱因或基础病史,症状无进行性加重,炎症及感染指标正常,支气管镜检查及病原学结果未见异常,病灶散在分布且以双上肺为主,并非以肺门为中心逐渐向外带扩散。
肺朗格汉斯细胞组织细胞增多症?支持点:青年男性,长期吸烟史,既往腹部皮疹病史,查体多发浅表淋巴结增大,HRCT双肺多发大小不等囊腔,形态不规则,以中上部肺野受累明显,支气管镜下穿刺活检提示慢性炎、炎细胞渗出及纤维组织形成;不支持点:肺活检标本及淋巴结活检标本未行免疫组化检查明确是否存在CD1a、S100表达阳性的朗格汉斯细胞,HRCT同时存在多发磨玻璃影。
肺淋巴管平滑肌瘤病也需排除。支持点:干咳、气短为主要临床表现,HRCT提示双肺多发囊腔;不支持点:青年男性,无显著的呼吸困难、咯血、自发性气胸等症状,囊腔分布于双肺中上部且大小不均一。
BHD综合征?支持点:HRCT双肺散在分布多发、大小不等的薄壁囊腔;不支持点:面颈部皮肤无纤维毛囊瘤,无肾脏肿瘤,家族史无异常,双肺囊腔分布无基底部胸膜下分布倾向。
还需鉴别是否感染性病变和恶性肿瘤。会是感染性病变吗?支持点:以干咳起病, HRCT提示双肺多发散在大小不等囊腔,右肺上叶后段存在混合密度结节;不支持点:无发热、咳痰,炎症及感染指标正常,肺泡灌洗液细菌、支原体、分枝杆菌等病原学检查均阴性。恶性肿瘤的可能呢?支持点:以亚急性干咳起病,左侧颈部淋巴结肿大,CT提示多发磨玻璃影、囊腔病灶,细支气管肺泡癌、转移性肿瘤可出现;不支持点:患者为青年男性,肿瘤标志物正常,肺泡灌洗液细胞学、支气管镜活检组织未查见癌细胞。
经过上述的鉴别诊断与分析,考虑先经验性抗感染治疗,同时密切观察和随访。
右肺上叶后段混合磨玻璃结节考虑感染性病变,戒烟后双肺囊腔改变逐渐缩小、减少甚至逐渐吸收,提示双肺囊性改变和吸烟密切相关
讲述者:傅恩清
紧接着我们给予口服莫西沙星片经验性抗感染治疗2周,嘱患者严格戒烟治疗。2021年11月18日,患者复查HRCT,结果见下图:
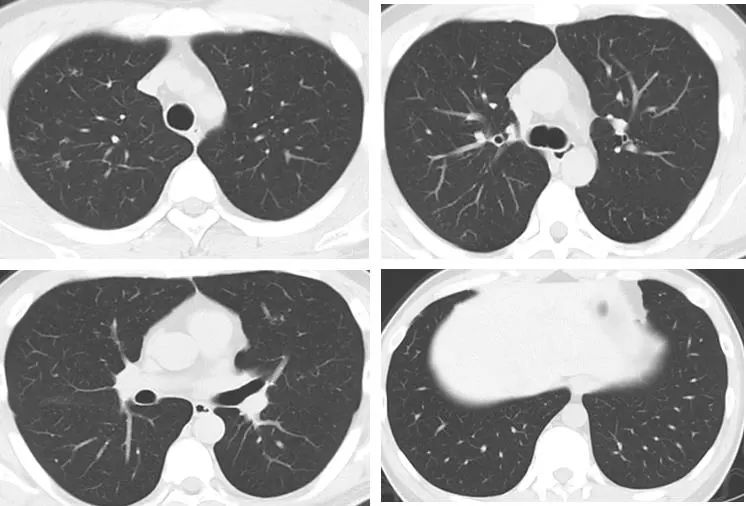
影像学显示,患者的右肺上叶后段混合磨玻璃结节较前明显吸收(小片状高密度影,边界不清,形态不规则,内可见直径约0.2cm实性结节影);双肺散在结节状、斑片状磨玻璃影;双肺多发囊腔较前减少,缩小。
2022年6月29日,患者到院复查胸部CT平扫,见下图:
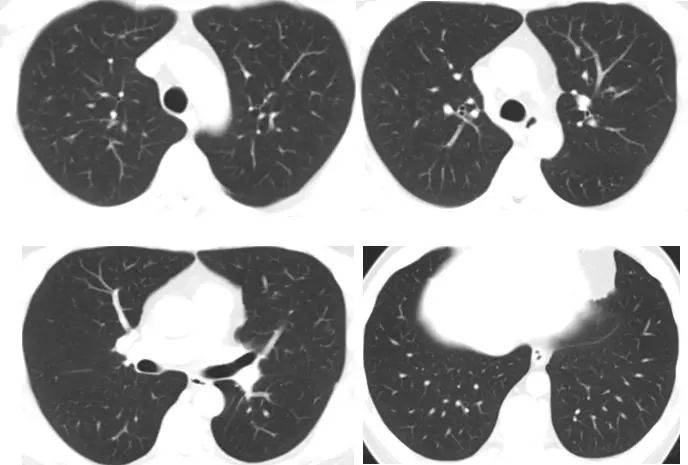
影像学显示,患者的双肺多发囊腔较前基本吸收,双肺散在结节状、斑片状磨玻璃影较前减少、缩小。根据患者的病史、体征及辅助检查结果,结合两次胸部CT随访结果,右肺上叶后段混合磨玻璃结节考虑感染性病变,严格戒烟干预下双肺囊腔改变逐渐缩小、减少甚至逐渐吸收,提示双肺囊性改变和吸烟密切相关,且戒烟有效,因此可以诊断为肺朗格汉斯细胞组织细胞增多症(pulmonary Langerhans cell histiocytosis ,PLCH)。
提及肺朗格汉斯细胞组织细胞增多症很多人会想到肺朗格汉斯细胞组织细胞增生症,两者之间有何区别呢?
朗格汉斯细胞组织细胞增生症(Langerhans cell histiocytosis ,LCH)是一种以CD1a+/CD207+未成熟树突状细胞(即朗格汉斯细胞,LC)异常增生,导致该细胞浸润组织气管和引起功能障碍为特征的克隆性、肿瘤增殖性疾病。LCH的好发部位依次是骨(80%)、皮肤(33%)、垂体(25%)、肝(15%)、脾(15%)、造血系统(15%)、肺(15%)、淋巴结(5%~10%)以及不包括垂体的中枢神经系统(2%~4%)[1]。
肺朗格汉斯细胞组织细胞增多症(pulmonary Langerhans cell histiocytosis ,PLCH)被定义为系统性LCH累及肺组织或单纯以肺组织为原发灶。该病起病隐匿,临床表现无特异性,误诊率及漏诊率较高。PLCH可发生于任何年龄段,高峰期为20-40岁,男女发病率几乎无差异。流行病学调查显示超过90%的成人PLCH患者有吸烟史[2]。此外,吸食大麻和PLCH的发生也有一定相关性。
该患者的特点更符合PLCH。目前PLCH的发病机制尚不清楚,既往研究认为单系统 PLCH是一种炎症/反应性障碍,因为它与吸烟或烟雾暴露几乎普遍相关,并且在戒烟后出现缓解[3]。但最新的研究[4, 5]发现约 86% PLCH患者检测到复发性 MAPK 通路改变,包括36% 至 50% 的病例中的BRAF -V600E,该观点也进一步支持了LC克隆性增殖的特征。此外,LCH伴发其他恶性肿瘤的概率很高[6],包括骨髓肿瘤(急性髓性白血病、骨髓增殖性肿瘤、慢性粒单细胞白血病)、淋巴瘤和实体肿瘤(肺癌和甲状腺癌),值得关注的是,PLCH伴发肺癌的机率也显著增高,可能有患者吸烟史相关[7]。
由于PLCH独特的发病机制和治疗方法,美国血液学会发布的最新版LCH诊治专家共识中将仅累及肺脏的LCH分类为单系统PLCH,将伴有肺外器官受累的LCH归为多系统LCH,以前者更为多见[8]。单系统PLCH临床表现多样,部分患者无症状;部分患者表现咳嗽、咳痰、活动后气促、呼吸困难、胸痛,咯血少见,10%-20%患者可出现自发性气胸。累及多系统的患者可出现肺外症状,如骨痛、皮疹、口干、多尿等。
作为低恶性程度肿瘤的PLCH,疑诊的重要手段是什么?目前治疗方案首选什么?通过该案例的诊治过程有何经验体会值得分享?
分析:臧宇
目前,高分辨率薄层CT(HRCT)是疑诊PLCH的重要手段,主要的影像征象如下:上中肺分布为主的囊腔影及结节影,累及肋膈沟附近的肺基底相对少见。疾病所处阶段不同,胸部 CT 表现也不尽相同。
一般认为,在疾病早期阶段,肺间质改变为主,特点是肺内网状阴影、小叶中心阴影增多、纵隔边缘不规则、叶间裂增厚及「磨玻璃」影,也可表现为双肺多发小结节影,结节大小不一,形态不规则,直径多在1-10mm,少数情况大于 1 cm。除典型结节影外,疾病早期阶段也可见类圆形囊状影。
随着疾病的进展,结节影可逐渐演变成空洞结节、囊肿,囊壁逐渐变薄。在疾病的终末期,囊肿之间不断融合,形成不规则的弥漫性囊腔、肺大泡,易发生气胸。若出现纵隔淋巴结肿大,需警惕合并恶性肿瘤。
值得注意的是,PLCH患者易合并肺动脉高压,出现呼吸困难的患者应行超声心动图评估。肺功能检查中以一氧化碳弥散能力下降最为多见(80%-90%),可同时伴有阻塞性、限制性或混合型通气功能障碍,约10%患者肺功能检查无异常。18F-FDG PET/CT可用于评估PLCH患者有无肺外器官组织受累、指导穿刺部位以及疗效评估,也有利于排除或尽早发现其他伴发恶性肿瘤[9]。
病变肺组织病理切片发现广泛增殖、浸润的朗格汉斯细胞是主要确诊依据,典型的瘤细胞呈片、巢状分布,胞质丰富、淡染,细胞核呈肾形、咖啡豆样、有核沟,背景常伴有淋巴细胞、嗜酸性粒细胞、组织细胞、纤维母细胞及多核巨细胞,特征性的免疫组织化学染色显示CD1a、CD207阳性是必要条件,其他常见的阳性指标包括S100、CD68和Ki-67,条件允许时可进行BRAF V600E的检测以指导治疗方案[1]。但经支气管镜活检组织病理诊断可能无异常或为炎症变化,支气管肺泡灌洗液检出率较低,肺泡灌洗液中CD1a阳性>3%时高度提示PLCH,当CD1a阳性>5%时具有诊断意义。当朗格汉斯细胞较少或难以找到时,可结合患者病史、患病初期的影像、肺部影像学的动态变化作出临床诊断。
目前针对PLCH没有统一的治疗方案,主要根据疾病累及范围和器官功能损害程度制订治疗方案。对于有吸烟史或被动吸烟的患者,严格戒烟为基础生活干预方案。有研究[2, 10]报道在孤立性PLCH患者中,部分患者仅通过戒烟可使病情稳定或完全缓解。
对于进行性肺功能恶化或影像学征象恶化进展的 PLCH 患者,有研究表明口服或静脉使用糖皮质激素具有一定的有效性[10, 11],但多为回顾性研究,无法排除戒烟对PLCH转归的作用,激素用法、剂量及疗程尚未统一,循证医学依据欠缺。有多系统受累及进展迅速的 PLCH 患者,化疗可为首选,但具体的化疗方案目前无统一规定,比较常用的化疗药有克拉屈滨、阿糖胞苷、长春新碱、依托泊苷、氨甲蝶呤等,具体的疗效存在个体差异[12]。MAPK 通路相关基因突变阳性的患者,靶向治疗是一种新的选择。BRAF激酶抑制剂 ,如维莫非尼(vemurafenib)、达拉非尼(dabrafenib)),MEK1 抑制剂,如曲美替尼(trametinib)[13],上述药物用于治疗 PLCH 或LCH取得了较好的效果。靶向治疗目前仍处于临床试验阶段,其长期安全性及有效性仍需要继续研究。对于肺功能持续恶化、肺实质破坏严重的晚期 PLCH 患者,肺移植是唯一的治疗手段,但移植后也存在一定的复发风险。
作为一种低恶性程度肿瘤,PLCH预后相对较好,但由于诊断时疾病所处阶段及对治疗反应的不同,个体患者预后差异较大,慢性呼吸衰竭和/或 肺动脉高压是导致患者死亡的主要原因,PLCH患者的10 年生存率为 93%,5 年和 10 年慢性呼吸衰竭和/或 肺动脉高压的累积发生率均<5%[14]。诊断后详细的全身评估、密切随访以及早期识别处理并发症的能够显著改善患者的远期预后。
该患者入院后我们给予了平喘、止咳治疗,使患者的干咳、气短症状明显改善,口服莫西沙星片经验性抗感染治疗2周后,再复查胸部CT提示右肺上叶后段混合磨玻璃结节较前明显吸收,双肺散在囊腔缩小、减少,双肺多发磨玻璃影无变化。经过严格的戒烟干预,患者8个月后再次复查胸部CT示双肺囊腔较前基本吸收,多发磨玻璃影较前减少、缩小,表明戒烟后患者病情完全缓解。总结本案的最后诊断及诊断依据请见第五份卷宗:
卷宗5
最后诊断
肺朗格汉斯细胞组织细胞增多症
诊断依据:
1.基本特征:青年男性,亚急性起病,有长期吸烟史。
2.症状:间断干咳2月,活动后气短3天,病情无明显加重或缓解。
3.体征:多发非对称性浅表淋巴结肿大,深部淋巴结及其他组织器官无异常、
4.辅助检查:
①胸部CT:双肺散在大小不等囊腔;双肺散在结节状、斑片状磨玻璃影;右肺上叶后段可见一混合磨玻璃结节,大小约2.3cm*2.3cm,形态不规则,边界不清晰,其内见直径约0.5cm实性结节影。
②入院血气分析、三大常规、生化指标、炎症感染指标、甲功、自身免疫相关指标均未见明显异常。
③肺功能、FeNO检测正常
④电子支气管镜下肺泡灌洗液及支气管镜保护性毛刷刷检均提示病原学检测阴性,未查见瘤细胞。
⑤电子支气管镜下观未见异常,EBUS-TBNB结果示右肺上叶后段支气管粘膜-肺组织慢性炎,伴炎性渗出及纤维组织增生。
⑥右侧腘窝淋巴结活检未见异常。
通过对本案例的诊治经过,团队的经验与体会如下:
1、对有吸烟史和典型肺部囊性病变表现者的鉴别诊断应将 PLCH 考虑在内。PLCH早期影像学表现不典型,以结节或空洞改变为主,且不同疾病阶段影像征象变化较大,应与多种间质性肺病、肺囊腔性疾病、肺气肿疾病相鉴别;
2、尽可能获取病变肺组织行病理形态及免疫组化检查明确是否为朗格汉斯细胞浸润,经支气管镜活检及肺泡灌洗液均有助于诊断PLCH,但后者检出率较低。若无法获取组织时可根据典型的影像学表现和病史做出临床诊断。首次诊断时应进行全面详细的体格检查、询问病史,有指征地选择相应辅助检查,必要时可取活检组织评估有无其他肺外组织器官受累;
3、单系统PLCH,除戒烟外,还可根据病情严重程度及进展速度选择随访观察、糖皮质激素、化疗等方案,密切随访及时识别并处理并发症有助于改善预后。戒烟后患者症状及HRCT征象缓解甚至完全消失有助于支持PLCH的诊断。
主任点评
肺朗格汉斯细胞组织细胞增多症是罕见的间质性肺疾病一种,也有认为是低恶性度肿瘤,发病率很低,但临床也有见到,诊断较为困难,病变肺组织病理切片发现广泛增殖、浸润的朗格汉斯细胞是主要确诊依据,本病例未活检到典型病例改变,虽然通过综合措施做出诊断,有条件应该进一步病理活检证实。本例患者治疗效果较好,结合临床做出诊断,初期即想到了PLCH诊断并进行了较为全面鉴别诊断,是较为成功病例,值得临床上学习。罕见病诊断常都较为困难,但应该想到,通过找依据,相关疾病的鉴别,也可以做出最终诊断,重要的是我们应该想到罕见疾病存在的可能性和特征是诊断的前提。
参考文献
[1] 中华医学会病理学分会儿科病理学组, 福棠儿童医学发展研究中心病理专业委员会, 中国抗癌协会小儿肿瘤专业委员会病理学组. 朗格汉斯细胞组织细胞增生症病理诊断专家共识[J]. 中华病理学杂志, 2022,51(08):696-700.
[2] Radzikowska E. Update on Pulmonary Langerhans Cell Histiocytosis.[J]. Frontiers in medicine, 2020,7:582581.DOI:10.3389/fmed.2020.582581.
[3] Liu H, Osterburg A R, Flury J, et al. MAPK mutations and cigarette smoke promote the pathogenesis of pulmonary Langerhans cell histiocytosis.[J]. JCI insight, 2020,5(4).DOI:10.1172/jci.insight.132048.
[4] Jouenne F, Chevret S, Bugnet E, et al. Genetic landscape of adult Langerhans cell histiocytosis with lung involvement.[J]. The European respiratory journal, 2020,55(2).DOI:10.1183/13993003.01190-2019.
[5] Mourah S, How-Kit A, Meignin V, et al. Recurrent NRAS mutations in pulmonary Langerhans cell histiocytosis.[J]. The European respiratory journal, 2016,47(6):1785-1796.DOI:10.1183/13993003.01677-2015.
[6] Bagnasco F, Zimmermann S Y, Egeler R M, et al. Langerhans cell histiocytosis and associated malignancies: A retrospective analysis of 270 patients.[J]. European journal of cancer (Oxford, England : 1990), 2022,172:138-145.DOI:10.1016/j.ejca.2022.03.036.
[7] Vassallo R, Ryu J H, Schroeder D R, et al. Clinical outcomes of pulmonary Langerhans'-cell histiocytosis in adults.[J]. The New England journal of medicine, 2002,346(7):484-490.
[8] Goyal G, Tazi A, Go R S, et al. International expert consensus recommendations for the diagnosis and treatment of Langerhans cell histiocytosis in adults.[J]. Blood, 2022,139(17):2601-2621.DOI:10.1182/blood.2021014343.
[9] 芮忠颖, 沈婕, 陆东燕, 等. 朗格汉斯细胞组织细胞增生症18F-FDG PET/CT显像一例[J]. 中国临床案例成果数据库, 2022(01):E624.
[10] 张莲华, 欧阳若芸. 15例成人肺朗格汉斯细胞组织细胞增生症临床分析及文献回顾[J]. 中南大学学报(医学版), 2022,47(03):334-343.
[11] Tazi A, de Margerie C, Naccache J M, et al. The natural history of adult pulmonary Langerhans cell histiocytosis: a prospective multicentre study.[J]. Orphanet journal of rare diseases, 2015,10:30.DOI:10.1186/s13023-015-0249-2.
[12] Vassallo R, Harari S, Tazi A. Current understanding and management of pulmonary Langerhans cell histiocytosis.[J]. Thorax, 2017,72(10):937-945.DOI:10.1136/thoraxjnl-2017-210125.
[13] Lorillon G, Jouenne F, Baroudjian B, et al. Response to Trametinib of a Pulmonary Langerhans Cell Histiocytosis Harboring a MAP2K1 Deletion.[Z]. 2018: 198, 675-678.
[14] Benattia A, Bugnet E, Walter-Petrich A, et al. Long-term outcomes of adult pulmonary Langerhans cell histiocytosis: a prospective cohort.[J]. The European respiratory journal, 2022,59(5).DOI:10.1183/13993003.01017-2021.
专家介绍

金发光
教授,主任医师,博士(后)导师,现任空军军医大学肺部疾病研究所所长、全军呼吸内科专科中心主任、国家呼吸内科医师培训基地、国家呼吸内科住院医师培训基地主任、陕西省呼吸疾病临床医学研究中心主任。兼任世界内镜医师协会呼吸内镜协会副会长、内镜临床诊疗质量评价专家委员会常务委员、国家肿瘤微创治疗产业技术创新战略联盟肺结节专业委员会副主任委员、陕西省医学会内科学分会主任委员、中华医学会内科学分会常务委员、全军呼吸内科专业委员会副主任委员、中国医师协会内镜分会常务委员和呼吸内镜专业委员会副主任委员、中国医师协会整合呼吸专业委员会主任委员,中国医师协会呼吸分会委员和呼吸内镜专业委员会副主任委员,《中华肺部疾病杂志》、副主编,《中华结核呼吸杂志》、《国际呼吸杂志》、《中国呼吸与危重症医学杂志》、《中国急救医学》、《中华诊断学杂志》编委。获国家、军队、陕西省各种奖项10项。承担国家重大专项、国科金、军队、陕西省各类课题20余项。共发表论文523篇,SCI收录78篇。

傅恩清
空军军医大学唐都医院呼吸和危重医学科,主任医师,教授,博士生导师。现任中华医学会呼吸病分会委员,陕西省医学会呼吸结核分会常委兼秘书,陕西省全科医师分会副主任委员,中华医学会呼吸病分会间质病学组委员,中国医师协会呼吸医师分会间质病学组委员,从事间质病临床诊断、治疗及发病机制研究。

臧宇
空军军医大学唐都医院呼吸与危重症科,副主任医师,陕西医师协会呼吸医师分会委员,陕西中西医结核康复专业委员会委员,陕西省保健协会呼吸专业委员,陕西医师协会睡眠专业委员,发表SCI及国内核心期刊学术论文10余篇,从事临床工作20余年,擅长呼吸系统疾病的诊断及诊疗,尤其擅长肺癌哮喘肺炎间质性肺病等疾病的诊治。
* 文章仅供医疗卫生相关从业者阅读参考
本文完
采写编辑:冬雪凝;责编:Jerry