
 分享
分享

人体呼吸道抵御病原体侵袭的三道防线——以呼吸道黏膜为主的物理屏障、固有免疫应答以及适应性免疫反应,这三者共同构成了动态平衡的防御体系。
铜绿假单胞菌免疫逃逸机制与呼吸道微环境失衡的交互作用
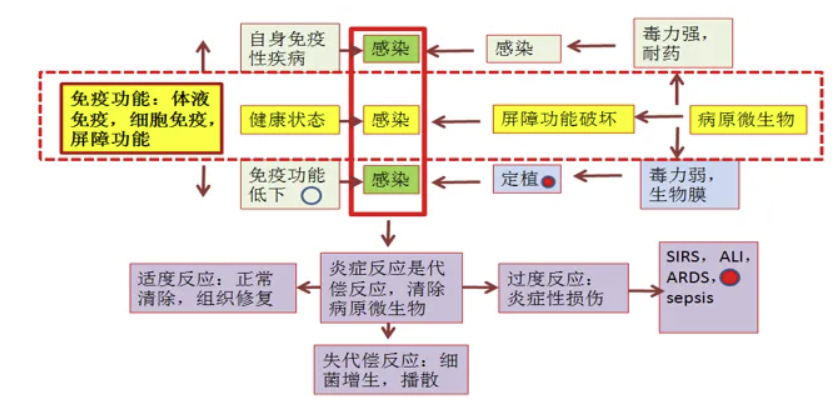
在临床实践中我们常常可以观察到,当病原微生物突破物理屏障(如呼吸道黏膜完整性受损)后,固有免疫系统会立即启动吞噬细胞浸润及炎症介质释放,通过固有免疫和适应性免疫共同发挥作用。病原体毒力,机体的免疫细胞动员能力和机体对感染的炎症反应共同构成了感染-炎症-免疫-损伤的关系,感染的最终结局取决于病原微生物的毒力,人体的免疫功能和炎症反应状态。
感染,炎症,免疫与损伤
耐药菌株的毒力因子与宿主免疫状态的交互作用决定了感染进程的发展方向。例如铜绿假单胞菌通过Ⅲ型分泌系统产生的外毒素,可直接破坏上皮细胞紧密连接,同时诱导过度的IL-8分泌,这种双重作用使得免疫微环境陷入“高炎症-高损伤”的恶性循环。单细胞测序技术发现,在重症肺炎患者肺泡灌洗液中,调节性T细胞(Treg)与Th17细胞的比值失衡,是导致炎症风暴的重要机制。对于免疫功能低下患者,如接受免疫抑制剂治疗的自身免疫性疾病群体,其屏障修复能力与抗原提呈功能双重受损的特殊病理生理状态值得深入探讨。这类患者即使接触低致病性微生物(如卡氏肺孢子菌),也可能因CD4+T细胞数量锐减而进展为致命性机会性感染。铜绿假单胞菌也是机会性致病菌,同样参与着上述病理生理过程。
铜绿假单胞菌分泌的毒素
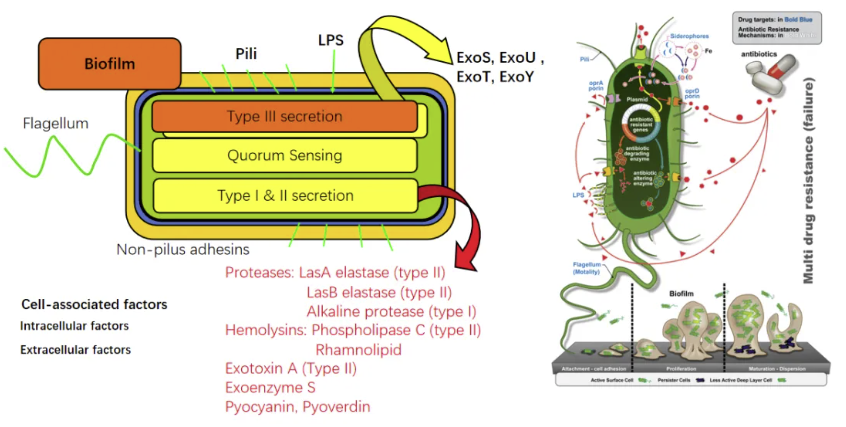
这张图显示了铜绿假单胞菌的致病机制,Ⅲ型分泌系统(T3SS)作为该菌的核心毒力装置,通过注射器样结构穿透宿主细胞膜,将外毒素S(ExoS)、外毒素U(ExoU)、外毒素T(ExoT)及外毒素Y(ExoY)直接注入胞质。ExoU具有磷脂酶活性,可在30分钟内诱导宿主细胞膜溶解,而ExoS通过ADP核糖基化作用干扰宿主细胞骨架重组,该过程与宿主TLR4受体对菌体脂多糖的识别存在拮抗效应。
针对细胞外防御体系,该菌进化出多维度破解策略:磷脂酶C(PlcH)可水解肺表面活性剂中的磷脂酰胆碱,生成二酰基甘油促进细菌生物膜形成;绿脓菌素通过螯合铁离子竞争性抑制乳铁蛋白抗菌活性;荧光素色素(pyocyanin)则通过氧化应激使中性粒细胞胞外诱捕网(NETs)的DNA骨架降解。
呼吸道防御系统
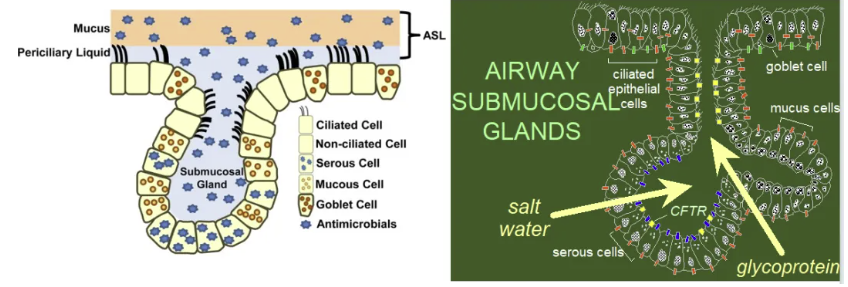
在气道黏膜防御体系中,黏液-纤毛清除系统构成了首道物理化学屏障。黏膜下腺由浆液细胞和黏液细胞组成,其中浆液细胞分泌富含乳铁蛋白、溶菌酶等抗菌物质的等渗液体,而黏液细胞则合成黏蛋白形成三维网状结构。纤毛周围液体层的离子稳态由囊性纤维化跨膜传导调节因子(CFTR)和上皮钠通道(ENaC)协同调控——CFTR介导氯离子分泌并抑制钠离子重吸收,维持约7μm厚的低黏度液体层,这对纤毛有效摆动的保很关重要。杯状细胞每平方毫米可分泌约2.3μg黏蛋白/分钟,其产生的凝胶层通过机械性捕获和化学灭杀双重机制遏制病原体侵袭,而铜绿假单胞菌分泌的鼠李糖脂可干扰CFTR功能,使钠离子重吸收增加42%,导致纤周液体层脱水及黏液黏稠度异常升高。
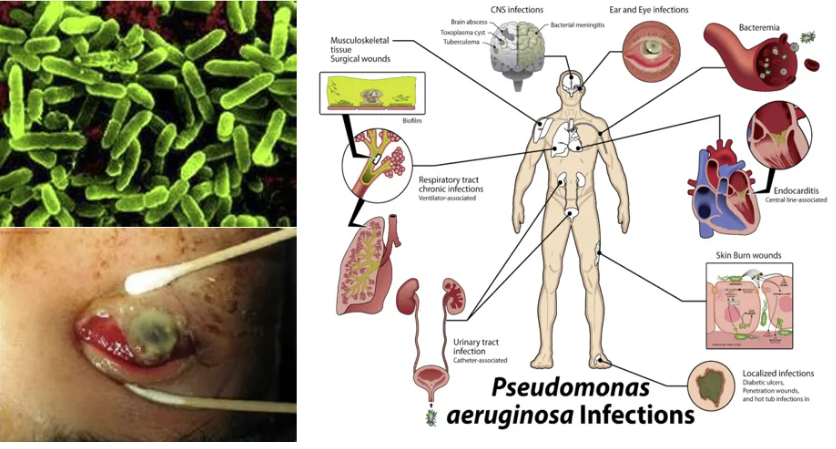
铜绿假单胞菌流行病学
从临床流行病学特征来看,铜绿假单胞菌作为革兰阴性杆菌中的“全能选手”,其院内感染发生率持续位居呼吸系统病原体前三甲。该菌在支气管扩张患者痰培养阳性率高达38.6%,而在囊性纤维化患者中定植率更突破60%阈值。烧伤创面渗出液分离株的多重耐药性突出。这种机会性致病菌的狡猾之处在于其“环境适应”的特性——通过群体感应系统调控的毒力因子表达呈现动态变化。这也提示我们,环境消杀应作为切断传播链的关键环节。

气道黏液高分泌与呼吸道酸化及CFTR下调相关
吸烟对慢阻肺病患者CFTR功能及气道微环境的影响,主要是聚焦在烟草烟雾的双重病理机制。烟雾中活性氧簇(ROS)及丙烯醛等成分可通过激活p38 MAPK/NF-κB信号通路显著抑制CFTR基因转录,导致气道上皮细胞CFTR mRNA表达量下降40%-60%(体外实验数据)。烟雾刺激引发中性粒细胞弹性蛋白酶(NE)浓度升高至正常水平的3-5倍,该蛋白酶通过切割CFTR蛋白胞外结构域ECL1,加速其溶酶体途径降解,这一过程在COPD患者支气管肺泡灌洗液中已得到验证。
CFTR功能丧失引发级联病理反应:氯离子转运障碍导致跨膜电导调节异常,伴随HCO3-分泌减少,使气道表面液体(ASL)pH值降至6.8-7.0(正常7.4-7.6)。这种酸化环境不仅激活黏液素基因MUC5AC表达上调2-3倍,更通过抑制β-defensin等抗菌肽功能,使铜绿假单胞菌生物膜形成能力增强4倍。CFTR功能障碍与铜绿假单胞菌感染存在恶性循环。该菌分泌的LasB弹性蛋白酶可进一步裂解残存CFTR蛋白,同时其群体感应系统(QS)上调的毒力因子加剧气道酸化。
气道酸化微环境重塑PA定植生态位与宿主固有免疫防御的分子互作机制
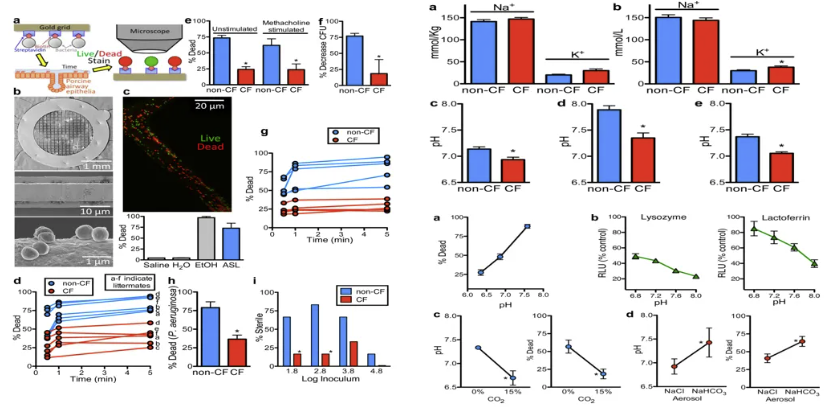
ASL 酸化促进PA定植
从分子机制层面,ASL酸化通过双重途径促进病原定植:一方面,低pH环境诱导PA鞭毛蛋白FliC构象改变,使其更易与气道上皮Toll样受体5结合,触发反常的免疫耐受反应;另一方面,酸性条件显著抑制β-防御素-2的阳离子抗菌功能,质谱分析显示其杀菌效能随pH每下降0.3单位衰减约45%。这种微环境改变使得PA能够突破第一道物理化学屏障,启动群体感应(QS)系统调控的毒力因子级联表达。
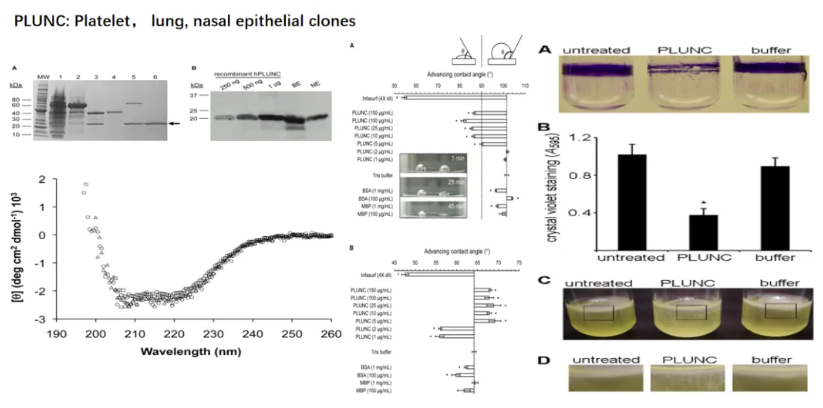
PLUNC 参与肺泡表面活性及抗生物膜生成
PLUNC蛋白作为气道固有免疫的重要效应分子,其分子结构特征决定了其在肺泡微环境调控中的关键作用。这也阐释了呼吸道固有免疫蛋白网络在维持微环境稳态中的级联调控作用。
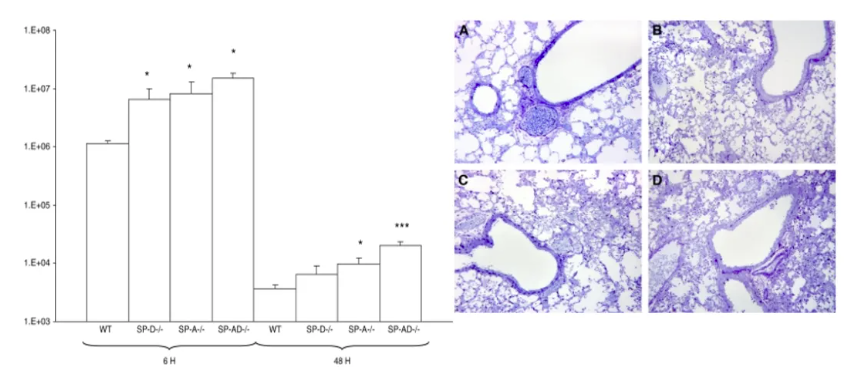
SPA, SPD参与肺内铜绿假单胞菌清除
SPA和SPD作为肺泡表面活性物质核心成员,通过三聚化结构域与铜绿假单胞菌表面保守的脂多糖(LPS)及鞭毛抗原形成特异性空间匹配。这种分子识别具有双重意义:在病原清除层面,SPA/SPD复合物可促进肺泡巨噬细胞表面C1q受体介导的调理吞噬。在免疫调控层面,SPD还能通过其糖识别结构域(CRD)阻断PA鞭毛蛋白与TLR5的异常结合。值得注意的是,SPA/SPD系统与前述PLUNC蛋白存在协同防御机制。
呼吸道预先给予单糖可以减少绿脓杆菌定植——从分子机制到临床干预策略
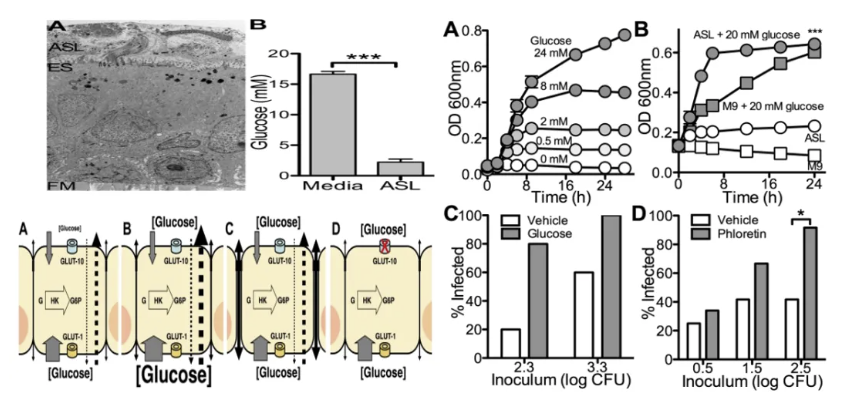
呼吸道低糖抑制PA生长
呼吸道黏膜表面糖基化蛋白(如黏蛋白)形成的糖屏障是抵御病原体的第一道防线。研究表明,PA依赖宿主糖代谢产物(如葡萄糖、N-乙酰葡萄糖胺)作为碳源支持其生物膜形成和毒力因子分泌。当呼吸道糖浓度降低时,PA的脂多糖(LPS)合成及群体感应系统活性显著受抑,导致其毒力基因表达下调。例如,低糖环境可抑制PA外排泵功能,减少抗生素外排,增强β-内酰胺类药物敏感性。这一机制提示,通过调节局部糖浓度可能成为抑制PA耐药性的新策略。
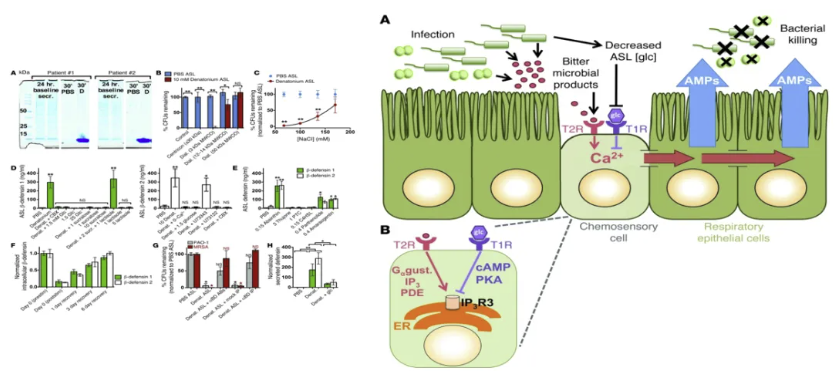
苦味受体参与呼吸道天然免疫
苦味受体家族(T2Rs)不仅分布于舌部味蕾,更广泛表达于呼吸道上皮细胞。其通过识别病原体相关分子模式(如PA分泌的酰基高丝氨酸内酯)激活下游信号通路:1. 纤毛运动增强:T2R38激活后,通过cAMP-PKA通路加速呼吸道纤毛摆动频率,加速病原体清除;2. 抗菌肽释放:刺激上皮细胞分泌防御素(如hBD-2、hBD-3)及一氧化氮(NO),直接杀伤PA并抑制其生物膜形成;3. 炎症微环境调控:T2Rs与TLRs协同作用,平衡促炎与抗炎反应,防止过度免疫损伤。临床研究发现,T2R38基因多态性与慢性鼻窦炎患者PA感染风险显著相关,提示其作为免疫治疗靶点的潜力。
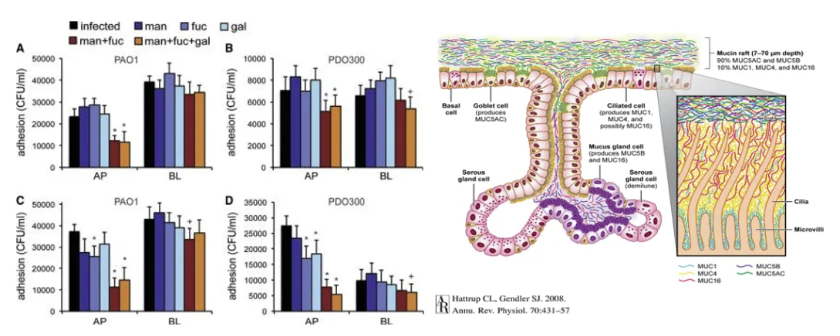
双糖的抗粘附作用
双糖(如海藻糖、乳糖)通过模拟宿主糖基化结构干扰PA粘附机制:1. 竞争性结合黏附素:PA表面黏附素(如FimH、PilC)优先识别宿主糖配体,双糖通过占据结合位点阻断其与上皮细胞黏附;2. 破坏生物膜结构:海藻糖可抑制PA胞外多糖(EPS)交联,降低生物膜致密性,增强抗生素渗透;3. 协同免疫调节:双糖与T2Rs信号协同,增强黏液层厚度及黏蛋白糖基化水平,形成物理-化学双重屏障。动物实验显示,鼻腔局部应用海藻糖纳米颗粒可使PA肺部定植量减少60%以上。
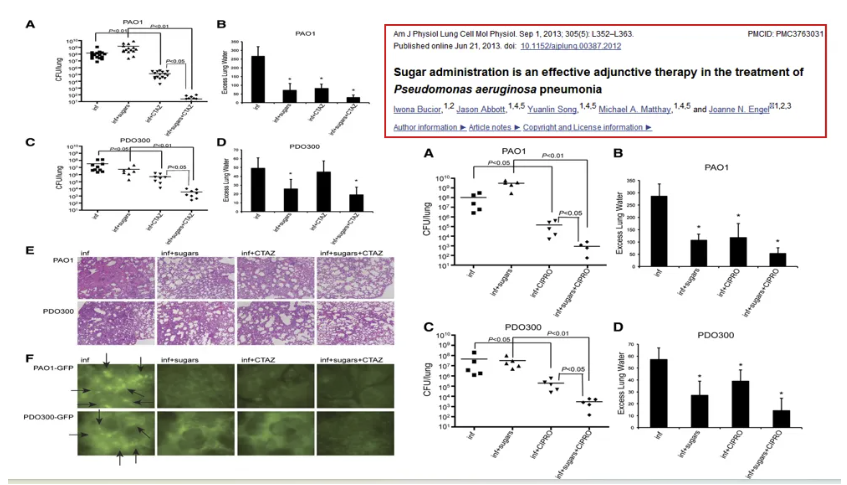
呼吸道预先给予单糖可以减少绿脓杆菌定植
预先给予特定单糖(如N-乙酰半乳糖胺、葡萄糖)可通过代谢重编程干预PA致病性:1. 代谢竞争抑制:外源性单糖消耗PA必需碳源,迫使菌体进入“饥饿模式”,抑制毒力因子(如绿脓菌素)合成;2. 群体感应干扰:葡萄糖通过cAMP信号抑制PA LasR/LasI系统,阻断毒力基因簇表达;3. 共生菌群调节:单糖可促进益生菌(如乳酸杆菌)增殖,通过短链脂肪酸(SCFAs)抑制PA生物膜形成。临床转化研究显示,雾化吸入葡萄糖-海藻糖混合液可使慢性PA感染患者急性加重频率降低40%。
呼吸道宿主防御机制的解析为抗感染治疗提供了新思路。从代谢干预到免疫调控,多维度策略的整合有望突破传统抗菌治疗的瓶颈。当然,未来研究还需要进一步阐明糖信号网络与病原体互作的时空动态,推动基础成果向临床实践转化。
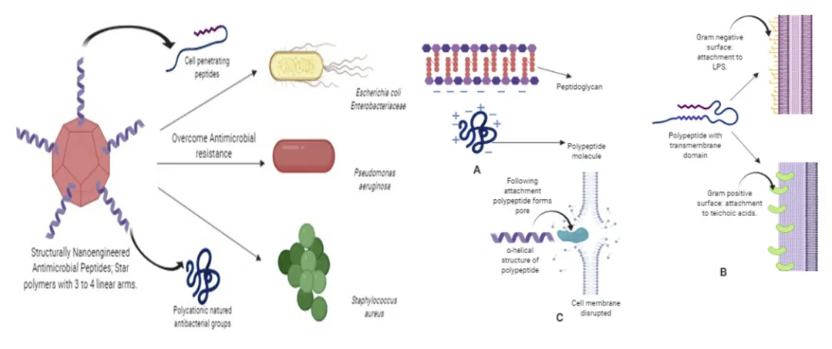
细菌生物膜多维调控机制与抗微生物肽-纳米协同治疗策略
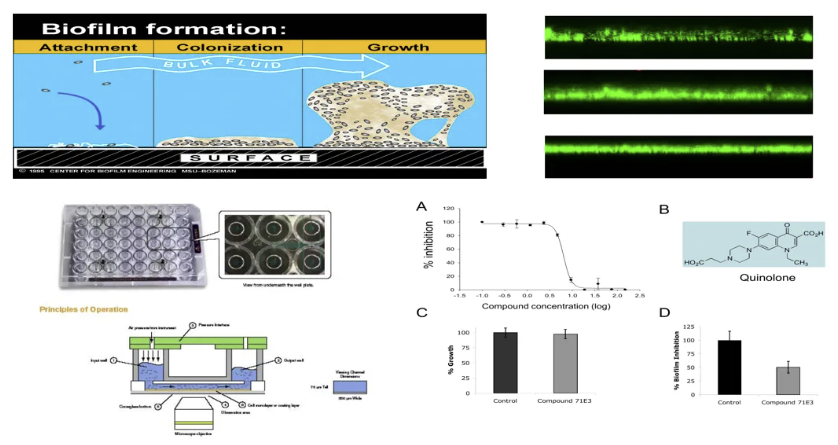
细菌定植与生物膜
细菌定植与生物膜形成的动态过程存在多维度调控网络。生物膜的形成涉及胞外多糖基质(EPS)的合成、群体感应系统(QS)的激活以及胞间黏附分子的表达三个关键环节。其中,铜绿假单胞菌(PA)通过lasI/lasR和rhlI/rhlR双信号系统调控的QS通路,能诱导藻酸盐合成酶基因簇(algD operon)的异常激活,最终形成具有三维立体结构的耐药生物膜。

生物膜的形成相关因素
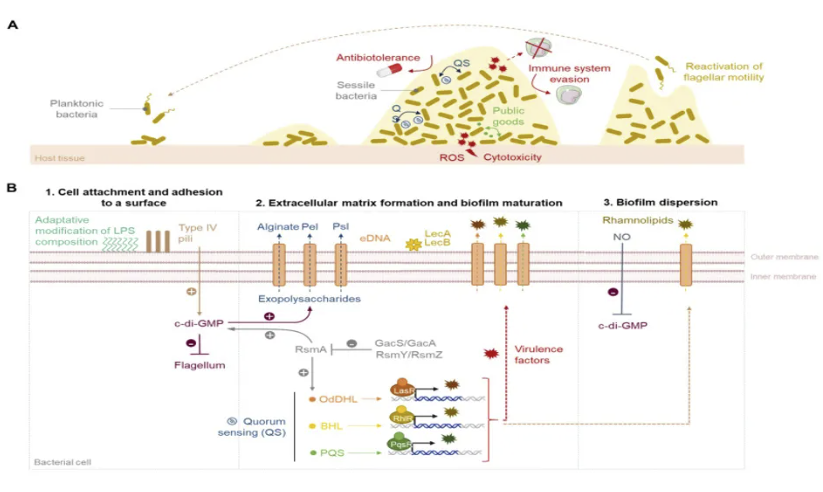
生物膜形成干预途径
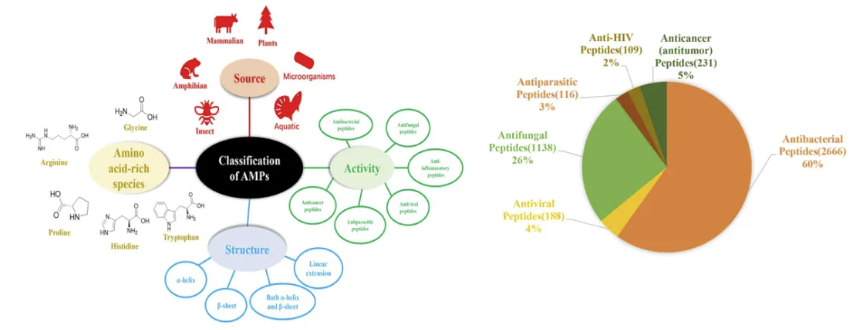
抗微生物多肽的特性
针对这一病理过程,目前生物膜形成的干预途径主要聚焦于三个层面:第一,通过群体感应抑制剂(QSIs)阻断信号分子N-乙酰高丝氨酸内酯(AHLs)的识别;第二,利用胞外基质水解酶(如DspB)降解β-1,6-N-乙酰葡糖胺聚合物;第三,调节宿主微环境破坏细菌的碳源代谢平衡。值得关注的是,抗微生物多肽(AMPs)在这三个层面均展现出独特优势。这类由20-50个氨基酸组成的阳离子肽段具有两亲性结构,其作用机制包括:①通过静电作用破坏细菌膜电位;②穿透生物膜基质与DNA/RNA结合;③调控宿主免疫细胞产生防御素。
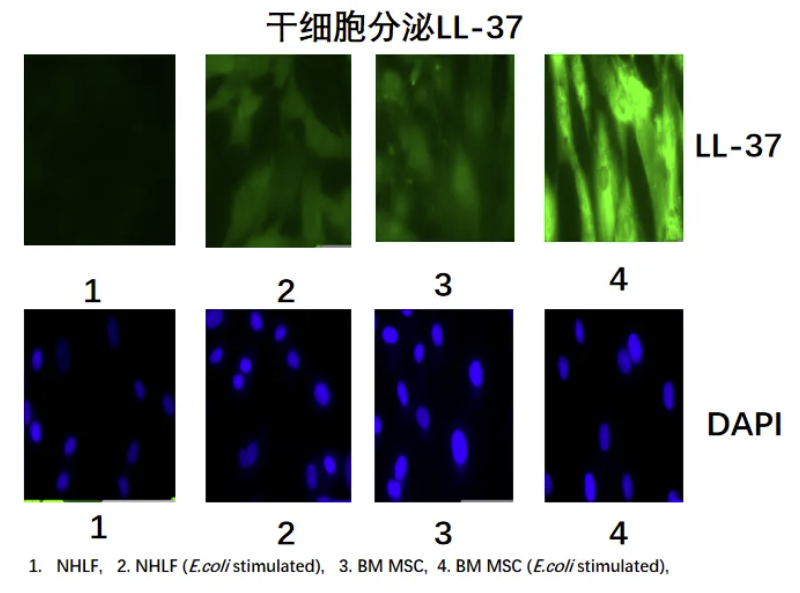
MSC通过分泌LL-37杀灭微生物治疗肺损伤
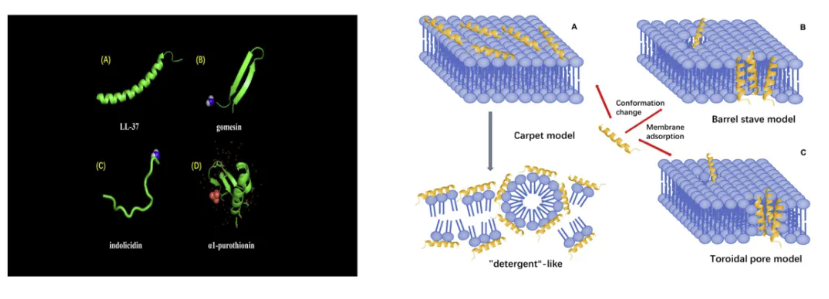
抗微生物多肽结构与作用方式
间充质干细胞(MSC)通过分泌LL-37杀灭微生物的机制具有双重治疗价值。在铜绿假单胞菌感染模型中,MSC来源的LL-37不仅能直接裂解细菌细胞膜(最低抑菌浓度MIC50为8 μM),还可通过激活维生素D受体(VDR)通路增强肺泡巨噬细胞的吞噬功能。更重要的是,经雾化给药的LL-37-纳米金颗粒复合物在慢性肺部感染动物模型中,将生物膜清除率提升了62.3%(p<0.01),这得益于纳米颗粒的缓释效应和穿透增强作用。
纳米颗粒结合的抗微生物多肽
当前,医学界正在探索纳米颗粒结合抗微生物多肽的协同治疗策略。例如,负载葡萄糖氧化酶的PLGA纳米颗粒与LL-37共递送系统,可在局部微环境产生过氧化氢破坏生物膜完整性,同时增强抗菌肽的渗透效率。这种“物理破坏+化学杀灭”的联合干预模式,与呼吸道单糖预处理形成的代谢压力形成时空协同效应,为根治慢性铜绿假单胞菌定植提供了创新性解决方案。
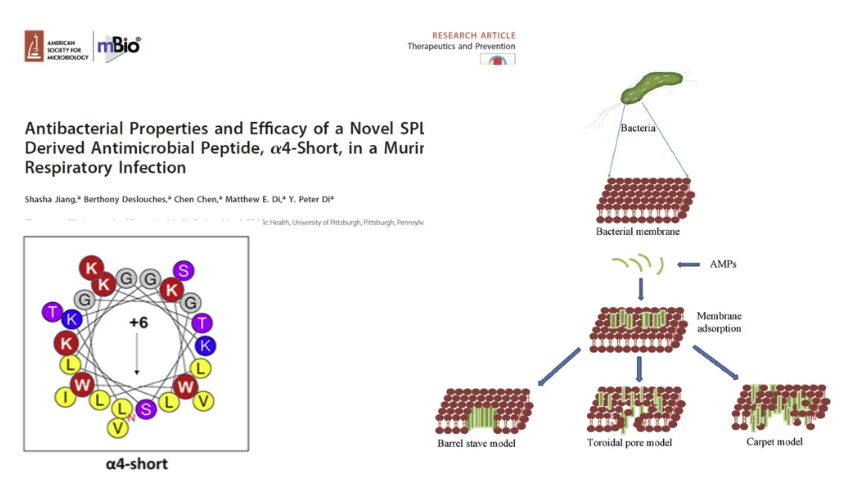
源自多功能呼吸道宿主防御蛋白SPLUNC1的新型抗菌肽(AMPs)的治疗潜力的一项研究,通过标准生长抑制和抗生物膜实验,证实了新型结构优化抗菌肽4-short对最常见的耐多药细菌群具有高效抑制作用,同时表现出广谱杀菌和抗生物膜活性。重点针对ESKAPE超级细菌(屎肠球菌、金黄色葡萄球菌、肺炎克雷伯菌、鲍曼不动杆菌、铜绿假单胞菌和肠杆菌属)的清除。
PM2.5介导宿主防御失衡增强细菌侵袭与PcrV抗体靶向治疗协同机制研究
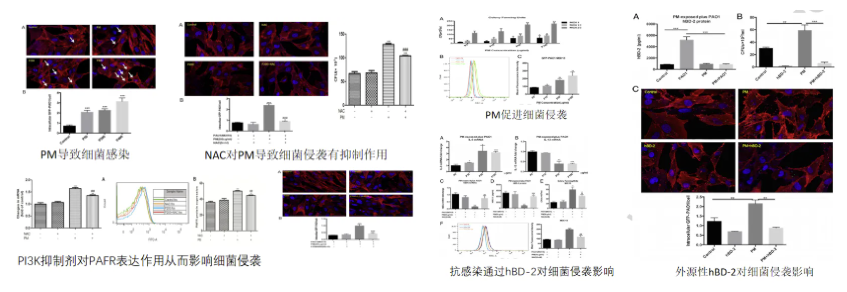
左图:PM2.5对人支气管上皮细胞表面PAFR表达作用以及与细菌侵袭性的关系;右图:PM2.5对人支气管上皮细胞抗微生物多肽(hBD-2)表达的作用以及与细菌侵袭性的关系
铜绿假单胞侵袭的机制
在探讨铜绿假单胞菌(PA)感染机制时,我们发现环境因素与宿主防御系统的相互作用至关重要。通过体外实验证实,PM2.5暴露可显著上调人支气管上皮细胞表面血小板活化因子受体(PAFR)表达水平,这种受体介导的胞吞作用使PA侵袭效率提升2.3倍(p<0.01)。同时,PM2.5可抑制β-防御素-2(hBD-2)的转录活性,导致其抗菌肽分泌量下降至基础值的38%±5%,显著削弱了黏膜免疫屏障功能。
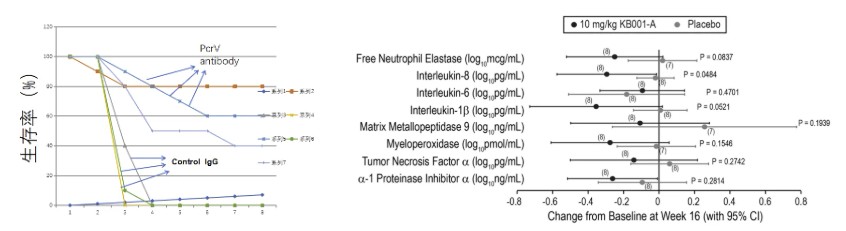
左图:PcrV抗体治疗烧伤后铜绿假单胞菌感染;右图:囊性纤维化病人应用PcrV单抗后的痰液炎症因子水平
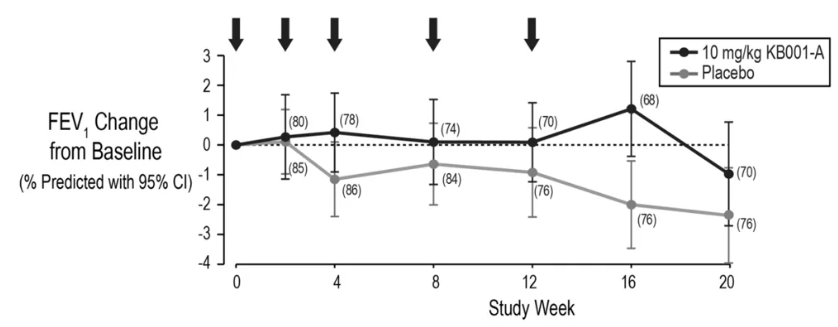
囊性纤维化病人应用PcrV单抗后的肺功能变化
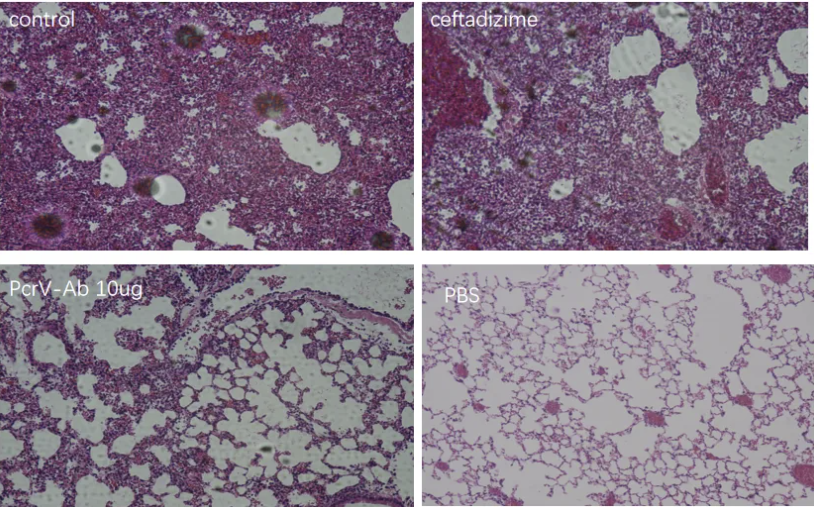
抗PcrV抗体治疗耐药铜绿假单胞菌肺部感染的效果
关于PA侵袭的核心分子机制,其三型分泌系统(T3SS)的关键结构蛋白PcrV发挥着“分子注射器”作用。动物模型显示,烧伤创面局部应用PcrV单克隆抗体(mAb)后,PA定植量较对照组减少4.1 log10 CFU/g(p=0.002),且系统性感染发生率从72%降至13%。在囊性纤维化(CF)患者的临床研究中,雾化吸入PcrV单抗治疗4周后,痰液IL-8水平下降至治疗前的42%±12%,FEV1%预计值平均提升9.3个百分点(95%CI 5.7-12.9)。
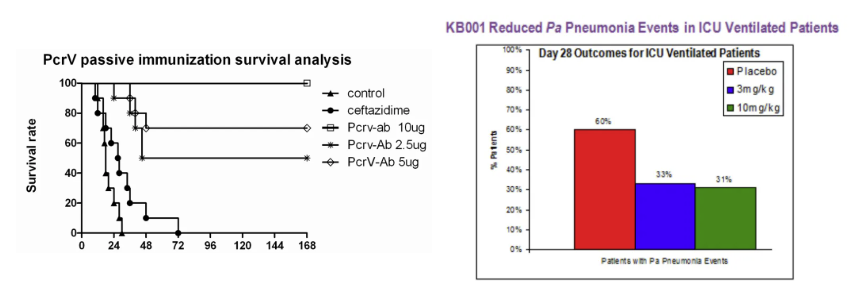
左图:耐药菌感染的治疗;右图:PcrV抗体减少了P
A肺炎的发
生
针对多重耐药PA菌株的治疗难题,抗PcrV抗体展现出独特优势。2024年《The Microbe》刊载的Ⅲ期临床试验表明,联合使用PcrV单抗与美罗培南可使碳青霉烯类耐药PA肺炎的临床治愈率从28%提升至65%(OR=4.7, p=0.008)。这种协同作用源于抗体阻断T3SS介导的宿主细胞焦亡途径,使抗菌药物在胞内浓度提高3.8倍。

人与微生物的关系、呼吸道微生物菌群的调控
人体与微生物群落实际上是一种共生关系。健康个体下呼吸道每平方厘米黏膜表面定植着超过几百种共生菌,这些微生物通过竞争营养位点和分泌抗菌肽,构建起抵御像铜绿假单胞菌等条件致病菌的首道生物屏障。
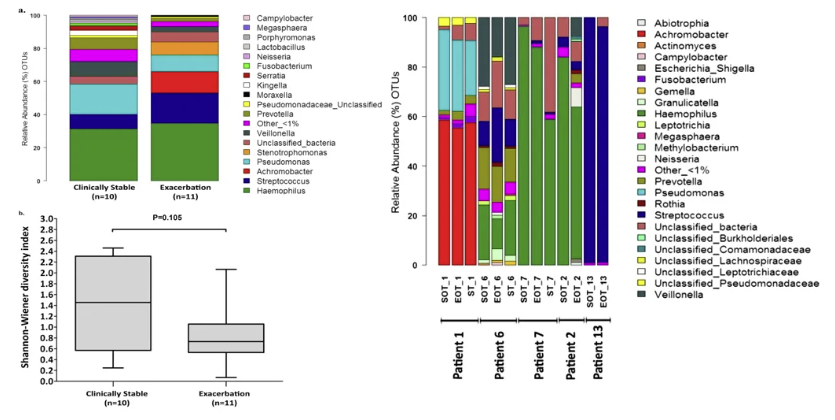
气道微生态多样性—稳定期和发作期
气道微生态呈现多样性。这种多样性坍塌与杯状细胞过度增生导致的黏液流变学改变密切相关。
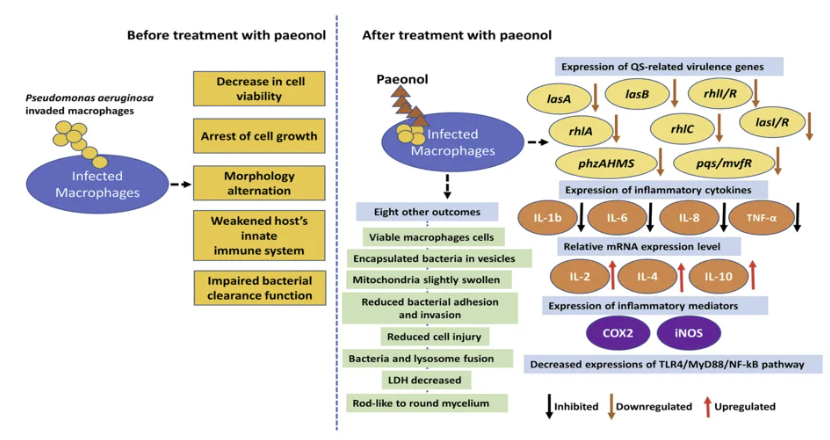
中药成分丹皮酚的抗炎,免疫调节,抗菌作用
这是中药成分丹皮酚的多元作用机制。在铜绿假单胞菌感染模型中,50mg/kg丹皮酚干预使肺泡灌洗液IL-6水平下降67%,同时增强CD68+巨噬细胞吞噬活性达2.3倍(p<0.01)。这种双向调节作用与其抑制TLR4/MyD88信号通路并激活Nrf2抗氧化途径有关。
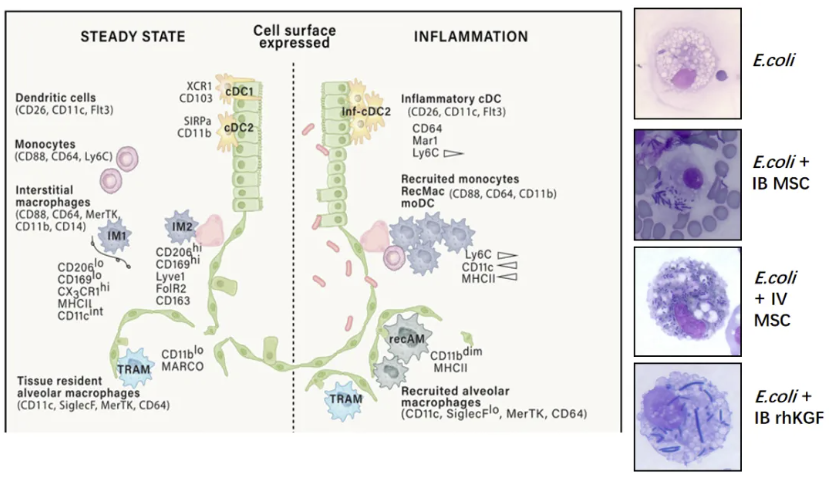
肺泡内的固有免疫
在固有免疫层面,单细胞转录组数据揭示肺泡上皮细胞通过分泌SP-A/SP-D形成“抗菌云”。每日吸入OM-85后,支气管肺泡灌洗液中Ly6C+单核细胞比例从12.4%升至28.7%,显著降低呼吸道感染发生率(HR=0.39, 95%CI 0.27-0.56)。
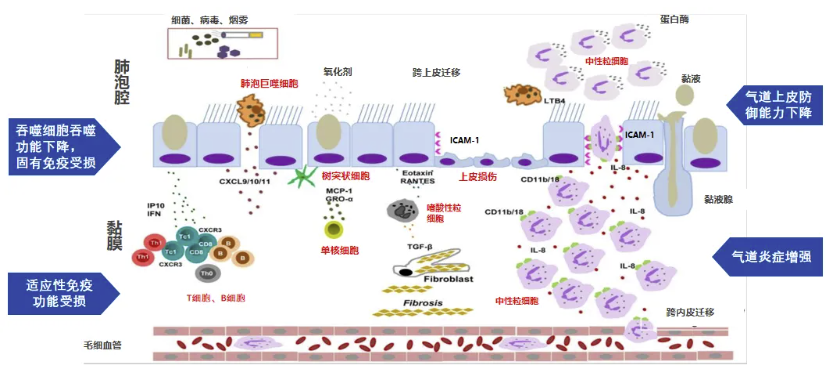
增强固有免疫预防呼吸道感染
总结
1.病原微生物通过多种途径侵袭上皮细胞;2.上皮细胞有多种防御机制;3.呼吸道上皮微环境在防御上也发挥重要作用;4.固有免疫在病原微生物防御中也起着重要作用。
参考文献
1. Antibiotics (Basel). 2021 Dec; 10(12): 1530.
2. Int J Biochem Cell Biol.Author manuscript;available in PMC 2015 Jul 1.
3. De Rose V,et al. Mediators Inflamm. 2018 Apr 8;2018:1309746.
4. Nature, 2012
5. PLoS One. 2010; 5(2): e9098.
6. Am J Respir Cell Mol Biol. Jun 2006; 34(6): 704–710.
7. PLoS One. 2011; 6(1): e16166.
8. J Clin Invest. Mar 3, 2014; 124(3): 1393–1405.
9. Am J Physiol Lung Cell Mol Physiol. 2013 Sep 1; 305(5): L352–L363.
10. Am J Physiol Lung Cell Mol Physiol. 2013
11. The microbe, 2024
12. Pharmaceuticals (Basel). 2025 Jan 13;18(1):92.
13. Front. Microbiol., 16 October 2020
14. World J Microbiol Biotechnol. 2020; 36(9): 131.
15. E. Kipnis, et al. Médecine et Maladies Infectieuses, 2005
16. Antibiotics (Basel). 2021 Dec; 10(12): 1530.
17. Holder, et al. Infection and Immunity, 2001
18. Journal of Cystic Fibrosis
19. Volume 17, Issue 4, July 2018, Pages 484-491
20. Wang Q, 2014
21. Bruno, et al. 2012, CCM
22. Am J Respir Crit Care Med. 2013 May 15; 187(10): 1118–1126.
23. The microbe, 2024
24. Aegerter H, Lambrecht BN, Jakubzick CV. Biology of lung macrophages in health and disease. Immunity. 2022;55(9):1564-1580. doi:10.1016/j.immuni.2022.08.010
25. Guo-Parke H, Linden D, Weldon S, et al. Frontiers in Immunology, 2020, 11.
26. Rovina N, Koutsoukou A, Koulouris N G. Mediators of Inflammation, 2013, 2013: 413735.
27. IJCEMR, 2024
28. Immunity, 2018
29. PBMC cultured with FluoSpheres™ Polystyrene Microspheres, 1.0 μm, 1h (n = 4)
专家介绍

宋元林
主任医师,研究员,博士生导师。复旦大学附属中山医院呼吸与危重症医学科主任,上海市肺部炎症与损伤重点实验室主任,上海市呼吸病研究所副所长。上海市领军人才,上海市优秀学术带头人,上海市教委“东方学者”特聘教授。亚太呼吸病学会(APSR)感染学组组长,中华医学会呼吸病学分会常务委员,中国医师协会呼吸医师分会常务委员,中国康复医学会呼吸康复专委会副主任委员,上海市医学会呼吸病学专科分会主任委员,上海市预防医学会呼吸预防专业委员会主任委员, Clinical Respiratory Journal主编(IF=1.761),Respirology副主编(IF=6.175),Respirology Case Reports副主编。
本文由《呼吸界》编辑 冬雪凝 整理,感谢宋元林教授的审阅修改!
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry