

 分享
分享

引言
当“罕见病”披上常见病的外衣,临床诊疗思路应从何入手?本案我们讲述了姐弟俩的病例,20年前,姐姐的死亡并非偶然,而是一种罕见病与临床认知局限共同作用的结果。20年后弟弟的确诊,不仅揭开了这种家族遗传病的死亡真相,更警示临床医生,当面对不明原因的多系统损害,尤其是合并乳酸酸中毒与呼吸衰竭时,需警惕这种疾病的可能。医学的进步,终将让“沉默的杀手”无所遁形。
当呼吸衰竭遇上乳酸酸中毒,是OSAHS还是代谢陷阱?
卷宗1:基本资料:患者女性,46岁,因主诉“胸闷、气促、下肢浮肿”于2005年5月23日入住我院。
讲述者:邱敬满

这是一张20多年前的很老旧的病历图。这位女患者于2003年8月无明显诱因出现双下肢、颜面浮肿及胸闷、气促。不能耐受一般活动,平卧时加剧,坐起后好转,无尿急、尿频、尿痛,无恶心呕吐,无胸痛、咳嗽、咳痰、咯血等,曾先后就诊于延安医院心内科及呼吸科院诊断为“先心病”,治疗后好转出院。于2004年两次住院治疗,病情稳定时生活能自理,但长距离行走或上一楼即感气促。入院前10余天症状加剧,入住我院心内科,在心内科住院期间,予强心、利尿、扩血管及氧疗治疗后;患者出现神志恍惚急查血气分析提示血气分析:pH 7.15↓, PO₂ 88mmHg↓, PCO₂ 120mmHg↑。予紧急气管插管治疗后PCO₂ 48mmHg;后患者自行拔管。后转诊于我院呼吸科;以“肺动脉高压原因待查、II型呼吸衰竭”收住。
入院查体:
T:36.8℃
R:25次/分钟
P:98次/分钟
BP:B110/80mmHg,一般情况差.吸氧状态下口唇中度发绀,平卧时颈静脉全程冲盈,双肺呼吸粗,双下肺可闻及少量湿性啰音,心界向两侧扩大,心率96次/分,律不齐,P2> A2,肝肿大,于剑下4cm,右助下3cm可触及,双下肢不肿。
完善相关检查:
2005年5月23日
血气:PH 7.27↓,PO₂ 41mmHg ↓,PCO₂ 84mmHg↑,BE 8.9,FI 21%。
血常规:WBC 10.44x10^9/L,Hb146g/L,血小板 138x10^9/L。
生化:肾功、电解质正常;乳酸 4.2mmol/L(0.7-2.1 mmol/L)
心电图:窦性心率,右心房负荷过重,T 波改变。
胸片:肺心病、肺动脉高压;右下肺野渗出性病变。
2025年5日17心脏彩超:右房右室扩大,,主肺动脉、右肺动脉增宽增宽,三尖瓣、肺动脉瓣轻度返流,左室收缩功能正常。中重度肺动脉压66mmHg。
(因年代久远,许多检查化验资料缺失)
患者呼吸科住院治疗2天后自动出院,出院2年后去世。20年后,她的弟弟又来到我院,以下是她弟弟的情况:
刘某某,男,59岁,汉族,离退休教师。主诉:运动不耐受40余年,呼吸困难伴睡眠打鼾3年余。
我们来看患者的病史过程:①患者40余年前出现长跑时呼吸困难,四肢无力,跑步距离下降,爬山、上坡费力;②出现左侧眼睑下垂,口角向左歪斜,伸舌左偏,左耳听力下降,考虑面瘫,半年后恢复;③3年余前出现轻微活动后呼吸困难伴睡眠打鼾,鼾声不大,夜间觉醒3-4次/晚,眠浅,晨起头昏,睡眠无恢复感,伴日间嗜睡,就诊于阜外心血管医院,完善相关检查,诊断“重度阻塞性睡眠呼吸暂停低通气综合征,呼吸衰竭、肺动脉高压、代谢性心肌病、线粒体病?”。建议佩戴呼吸机治疗,患者拒绝。现患者呼吸困难、心悸加重特来我科就诊。
既往史:4年前患者出现胃痛、反酸,考虑胃食管反流,2年前因心动过速,行心脏射频消融术。无嗜烟、嗜酒史。无手术史。发育情况:妊娠期无特殊,足月出生,出生过程正常,婴幼儿期语言、行为发育正常。家族史:母亲40岁时因直肠癌手术3天后去世,去世前未发现类似症状,父亲68岁因肺心病去世。1姐体格瘦小,跟患者有相似病史,运动不耐受、呼吸困难多年,高二氧化碳昏迷多次,死于不明原因呼吸衰竭,48岁去世。否认家族中其他人有类似病史,否认有近亲婚配史。
一般情况可,神清,生命征平稳,身高:164cm,体重:48kg,BMI:17.84Kg/㎡,身材瘦小,左耳听力减退,口唇轻度发绀,咽腔稍狭窄,扁平胸,呼吸音稍低,未闻及干湿啰音,未闻及胸膜摩擦音。心腹未查及异常。双下肢无水肿。颅神经未见明显呼吸异常,双下肢曲髋肌力4+级,余肢体肌力5级,四肢腱反射减弱,病理征未引出,感觉、共济无异常,脑膜刺激征阴性。
精神检查:意识清醒,定向正确,接触好,应答切题,未引出明显精神病性症状,情感适切,自知力存在,社会功能正常。
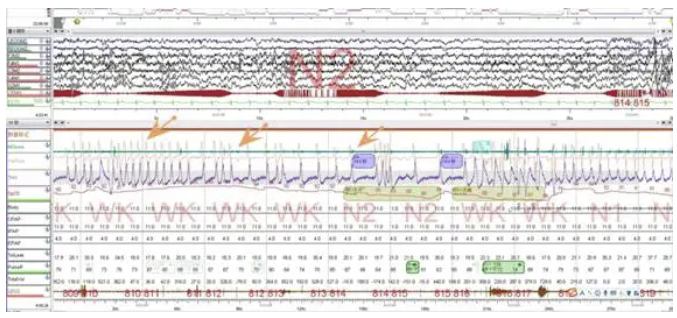
入院前检查:PSG:1.重度阻塞性睡眠呼吸暂停低通气综合征;2.夜间重度低氧血症。AHI 46.5次/小时;阻塞性低通气事件为主;最低血氧饱和度30%。
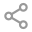
肌肉活检电镜检查:

肌肉活检病理检查:
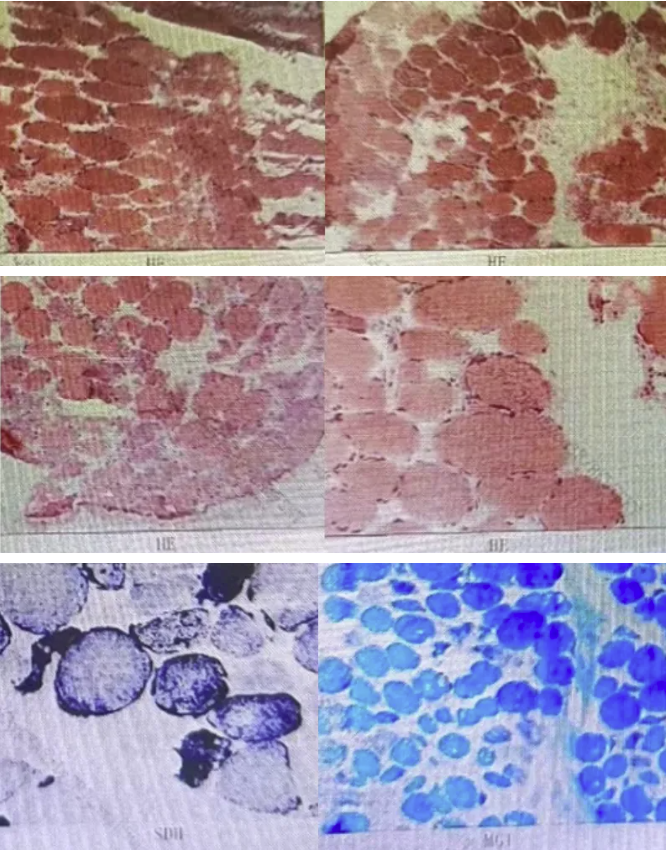
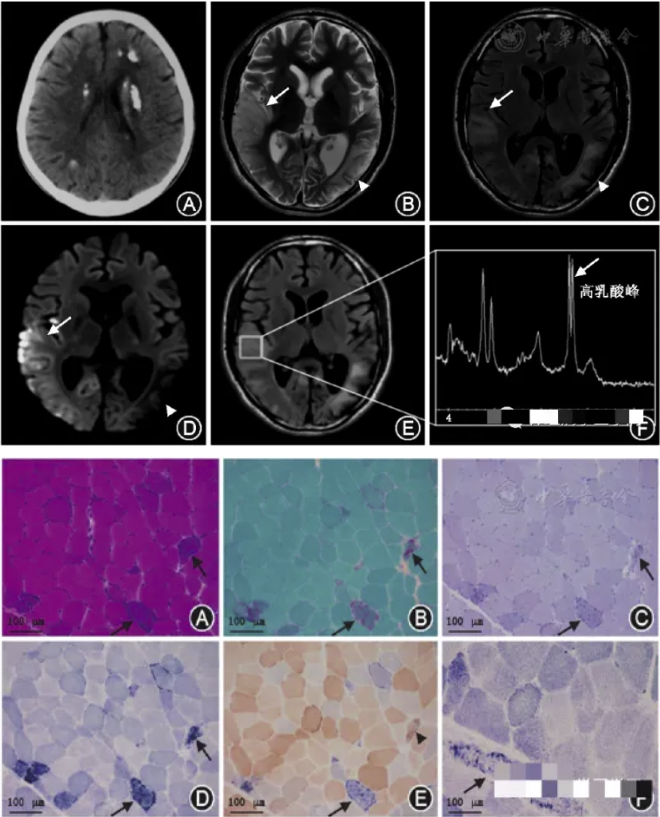
2021年于外院心血管内科:生化测定:LAC 8.08.0mmol/L↑,肌酸激酶124 U/L↑,肌酐41umol/L↓。肌肉活检:可见RRF/破损蓝纤维,电镜见晶格状线粒体等异常线粒体。
初步诊断:1.阻塞性睡眠呼吸暂停低通气综合征;2.呼吸衰竭;3.代谢性心肌病;4.肺动脉高压;5.返流性食管炎;6.高尿酸血症;7.左耳听力下降;8.线粒体病?入院后积极完善相关检查:肌酸激酶升高,肝肾功、电解质、心肌损伤标志物、血常规、凝血功能、甲功、血流变、大小便常规、ANCA、ANAS、抗心磷脂抗体谱均未见明显异常。
心脏彩超:左室壁增厚、肺动脉压45mmHg(轻度升高)。动态脑电图:正常。四肢肌电图:正常。腹部B超、头颅MRI未见明显异常。心电图正常。
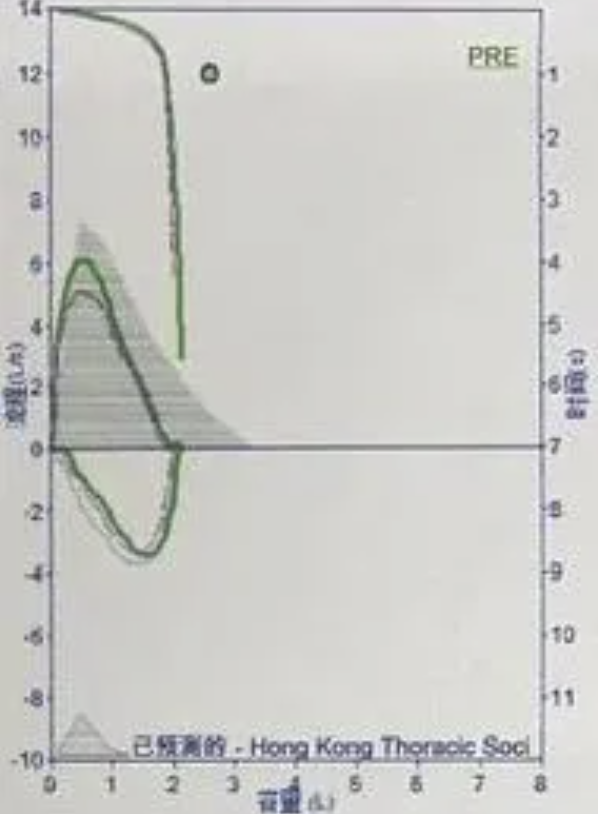
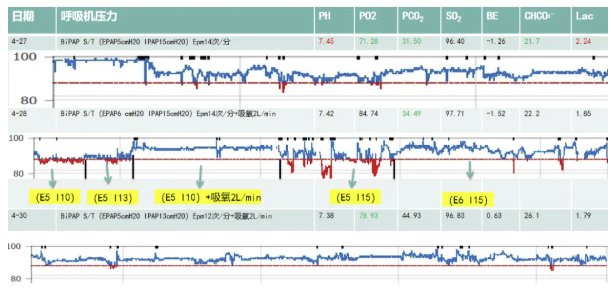
呼吸功能相关检查:呼吸中枢低氧驱动、高二氧化碳驱动功能均低于实验正常值。 喉镜:鼻中隔偏曲,咽炎。(2023-04-22)日间血气:PH 7.37↓,PO2 57mmHg ↓,PCO2 47mmHg↑,SO2 88↓BE 1.25,CHO3- 27.2↑,LAC 5.91 mmol/L↑。(2023-04-26)肺功能报告(立位):肺通气功能正常,弥散功能正常。肺功能报告(卧位):轻度限制性通气功能障碍。
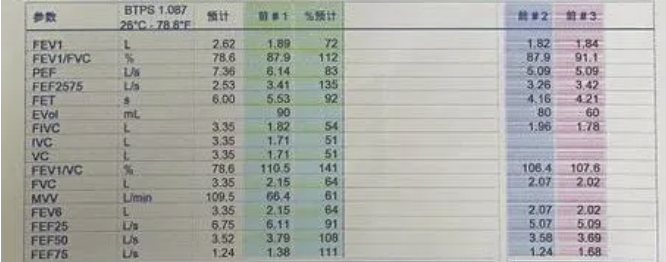
PSG监测:
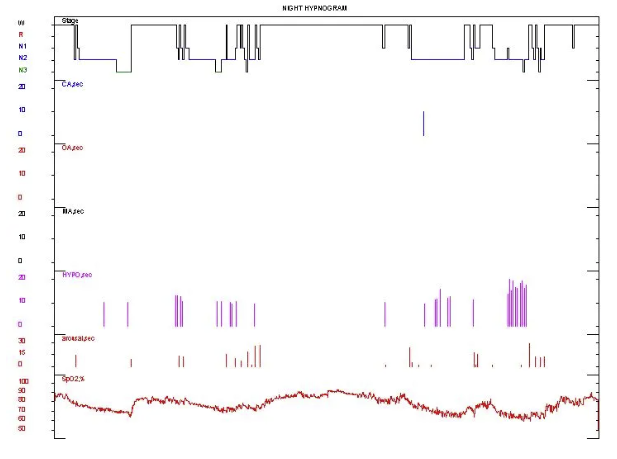
中度阻塞性睡眠呼吸暂停低通气综合征的PSG表现。以阻塞性低通气为主,AHI23.8次/h。最低血氧饱和度 59%。
监测结束后血气:PH 7.34↓,PO2 49mmHg ↓,PCO2 57mmHg↑,SO2 83↓ LAC 1.821↑
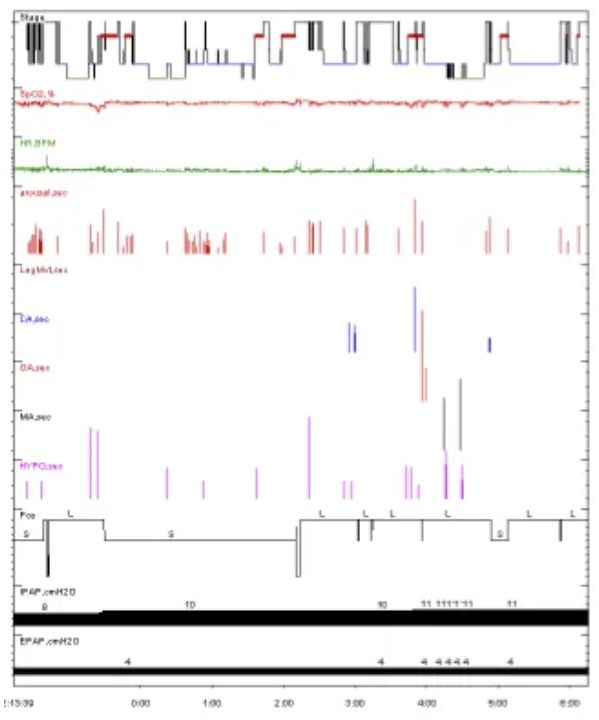
人工压力滴定:脑电大致正常。滴定压力BIPAP S/T(E:6,I:14)cmH20,T12次/分。
现在,我们将这个弟弟的情况大致做一个总结:患者刘某,男,59岁,离退休教师。主诉是“运动不耐受40余年,呼吸困难伴睡眠打鼾3年余”。他有几段关键病史:40年前长跑时呼吸困难,伴左侧眼睑下垂、面瘫(半年后恢复);3年前活动后气促、睡眠打鼾,外院诊断“重度OSAHS、呼吸衰竭、肺动脉高压、代谢性心肌病、线粒体病?”家族史:姐姐(48岁死于“不明原因呼衰”)、母亲(40岁术后猝死),均有运动不耐受史。他的入院检查,注意体征:身材瘦小(BMI 17.84),左耳听力减退,口唇发绀,扁平胸。另外有几个关键异常值:乳酸(LAC):8.08 mmol/L↑(外院);血气分析:pH 7.34↓, PO₂ 49mmHg↓, PCO₂ 57mmHg↑, LAC 1.82 mmol/L↑。
患者是以“呼吸衰竭+OSAHS”就诊的,但以下几个矛盾点需要我们思考和深究:1.为何乳酸持续升高?感染/休克已排除,需考虑代谢性疾病;2.为何年轻姐姐猝死?家族史高度提示遗传病;3.为何卧位肺功能异常?提示呼吸肌受累。
20年谜案终破解……从“肺动脉高压”到MELAS的惊心跨越
讲述者:刘畅
治疗上,给予无创正压通气。
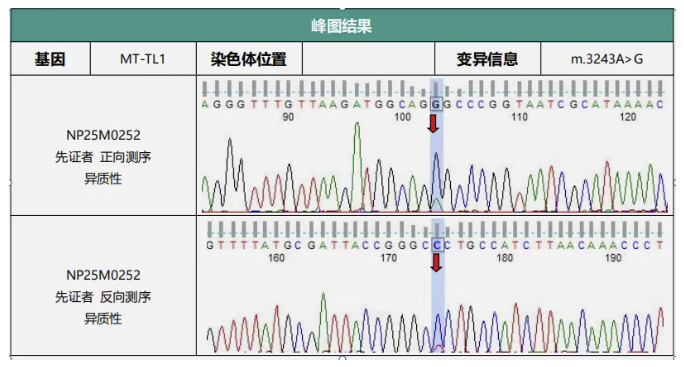
线粒体基因组全长检测:

检测到了可以解释患者表型的致病变异。

诊断:线粒体脑肌病伴乳酸血症和卒中样发作(MELAS)。至此,明确诊断为:
1.呼吸衰竭
2.睡眠相关肺泡低通气障碍
3.阻塞性睡眠呼吸暂停低通气综合征
4.线粒体脑肌病伴乳酸血症和卒中样发作(MELAS)
5.代谢性心肌病
6.肺动脉高压
7.返流性食管炎
8.高尿酸血症
9.左耳听力下降
10.肺部阴影
出院医嘱:1、夜间规律佩戴呼吸机BiPAP(EPAP 5cmH2O,IPAP 13cmH2O,T 12次/分+吸氧2L/min;2、长期服用辅酶Q10及左旋精氨酸
患者2023年6月11日复诊:
清醒状态下不吸氧,氧饱可达到93%。
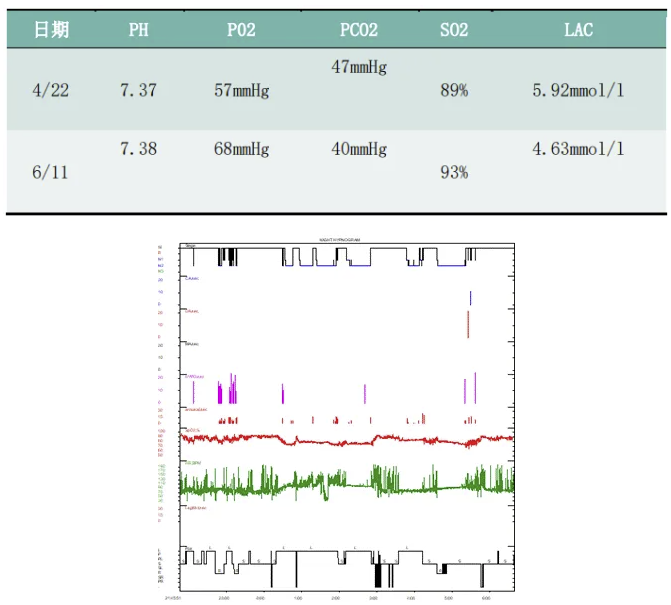
1、睡眠效率降低,深睡眠缺失,R期睡眠缺失。
2、符合轻度阻塞性睡眠呼吸暂停低通气综合征的PSG表现AHI 7次/小时,以低通气为主。
3、睡眠中平均氧饱和度83 %,最低氧饱和度66%。
患者压力滴定时出现,多次因呼吸肌无力造成的无效触发。
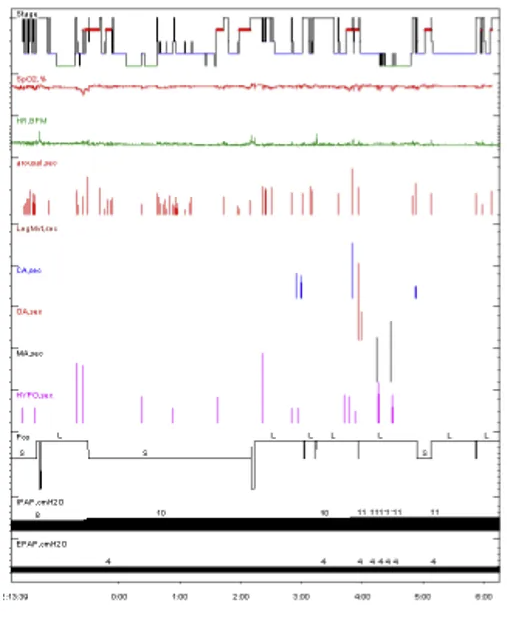
最终滴定压力BIPAP ST(E:4,I:11)cmH2O。
从以上我们可以整理出一条诊断逻辑链:1.初始误诊(2005年姐姐):姐姐是以“胸闷、气促、浮肿”就诊,误诊为“先心病”,按“原发性肺动脉高压”治疗无效死亡。漏诊点是未深究“运动不耐受40年+反复高碳酸血症昏迷”的遗传背景。2.弟弟的再认识(2021年):患者的多系统损害,运动不耐受+面瘫+听力下降+心肌病。呼吸特征:呼吸中枢对低氧/高CO₂敏感性降低(化学驱动障碍);REM睡眠期严重低氧(膈肌电活动异常)。金标准证据,一是肌肉活检:RRF纤维+异常线粒体,二是基因:mtDNA 3243A>G突变(MELAS致病突变)。
在这两个案例的鉴别诊断当中,最初姐姐被疑似诊断为特发性肺动脉高压,但姐姐的尸检是缺失的,但从弟弟的肺动脉压仅轻度升高(45mmHg),且合并多系统损害,可以发现端倪。其中还有一个关键突破点:乳酸酸中毒+呼吸中枢驱动障碍的组合,直指线粒体能量代谢衰竭。
关于MELAS的呼吸困局,我们该如何避开“代谢的陷阱”?从治疗上其实是有所启示的。1.呼吸支持特殊性:无创通气模式:BiPAP S/T模式;参数设置:低压力支持(IPAP 11-13cmH₂O)+ 高触发灵敏度(防呼吸肌乏力);氧疗策略:夜间低流量吸氧(2L/min)纠正缺氧,避免乳酸堆积恶化。2. 代谢干预核心:辅酶Q10+左旋精氨酸:改善氧化磷酸化,急性期降颅压;避免乳酸蓄积:纠正酸中毒(碳酸氢钠慎用,加重细胞内酸中毒);3. 家族筛查必要性:姐姐女儿需基因检测(母系遗传风险50%)。
这里能给呼吸科医生几个警示,首先我们要警惕“伪装者”乳酸酸中毒:呼吸衰竭患者若合并顽固性高乳酸血症(>3mmol/L),需跳出感染/休克思维,筛查代谢病;其次是关注“反常”的呼吸参数:卧位限制性通气障碍+REM期重度低氧,提示呼吸中枢或膈肌受累(神经肌肉病标志);第三是需要深挖家族史,不明原因青年猝死+多代运动不耐受=遗传性代谢病高危信号。
对于神经肌肉疾病导致肺泡低通气的患者,无创通气需采用BPAP(S/T)模式,需设置较高的吸气触发灵敏度,降低因呼吸肌无力造成的无效触发。通常不需要较高水平的压力支持,并且对高水平的压力支持耐受性较差。
线粒体脑肌病伴乳酸血症和卒中样发作(MELAS),是由于mtDNA或核DNA突变,引起线粒体呼吸链氧化磷酸化结构和功能障碍为特点的一组遗传代谢疾病。以卒中样发作、癫痫样发作、身材矮小、心肌病、糖尿病、高乳酸血症、肌肉疲劳无力为主要临床特点。是线粒体脑肌病中最常见类型。多为母系遗传。[1]
病因方面:约80%的突变为mtDNA 3243A>G突变;mtDNA突变,致转录终止,无法编码氧化代谢过程中必需的酶/载体,致糖原/脂肪酸等底物不能进入三羧酸循环,不能被充分利用。致ATP产生不足,最后导致细胞功能减退甚至坏死;突变基因必须超过临近极限才能产生临床症状,更易累及能量需求高的器官或组织(脑、心肌、骨骼肌),偶见呼吸肌受累。[2-3]
该病临床表现:任何年龄均可发病,发病高峰在10~30岁。青少年卒中样起病,有偏头痛,智力低下,身材矮小,神经性耳聋,反复癫痫发作,常有不耐疲劳,可有阳性家族史。其他系统:心脏系统:心肌病和充血性心衰,以扩张型、肥厚型心肌病为多见,患者有心脏传导异常也很常见。内分泌系统:糖尿病、糖耐量异常。消化系统:便秘和胃痛,偶见反复发作的恶心、呕吐、胃肠麻痹、胰腺炎。以及可有腹泻病。
辅助检查:1.生化测定:血清肌酸激酶正常或增高,肌酸激酶/乳酸脱氢酶比例倒置,血乳酸升高;2.头颅影像学:影像学特征性改变。为非单一血管支配的病灶,具有进展性、可逆性、游走性特点;3.电生理检查:肌电图、脑电图;4.组织病理学检查:骨骼肌活检冰冻切片的典型病理改变是改良Gomori三色染色可见不整红边纤维(RRF),琥珀酸脱氢酶染色可见破碎蓝染肌纤维和(或)深染的小血管;5.基因检测:mtDNA基因突变分析是诊断该病的最敏感方法。
鉴别诊断:脑小血管炎、心源性脑栓塞、大脑皮质静脉血栓形成、病毒性脑炎、自身免疫性脑炎、甲基丙二酸血症、高氨血症、癫痫后脑部MRI可逆性信号改变、可逆性后部脑病综合征等。
治疗目标:维持人体能量代谢平衡以提高患者生活质量
1.代谢治疗:艾地苯醌和辅酶Q10是治疗氧化磷酸化缺陷的首选药物。生酮饮食、运动康复训练;2.卒中样发作急性期治疗:L-精氨酸;3.对症治疗;4.基因治疗。
下面我们来谈谈线粒体脑肌病伴卒中样发作(MELAS)相关呼吸功能障碍的病理生理机制可概括如下:
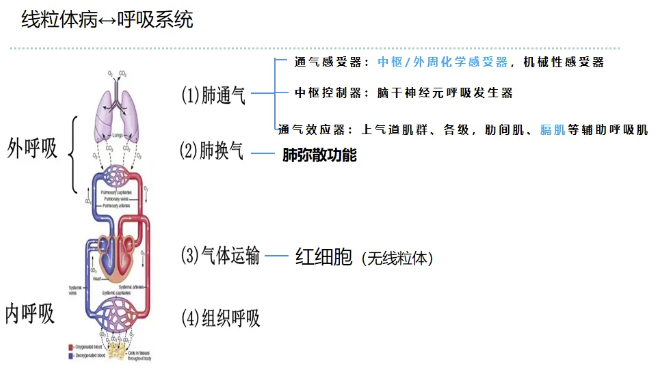
1. 内呼吸与气体运输障碍
内呼吸包含气体在血液中的运输及组织呼吸(细胞内氧化磷酸化)。虽然成熟红细胞无线粒体,但其通过Hb构象变化完成氧与二氧化碳的运输为主动耗能过程,依赖ATP供能。线粒体功能障碍导致能量代谢异常,可影响红细胞的气体运输效率。同时,组织呼吸受损,氧化磷酸化效率降低,乳酸生成增加,加剧代谢性酸中毒。
2. 外呼吸功能障碍的评估与机制
外呼吸包括肺通气与肺换气。该患者肺弥散功能(DLCO)正常,提示肺换气环节(肺泡-毛细血管膜气体交换)受疾病直接影响较小。功能障碍主要集中于肺通气环节,其调控与执行涉及以下部分:
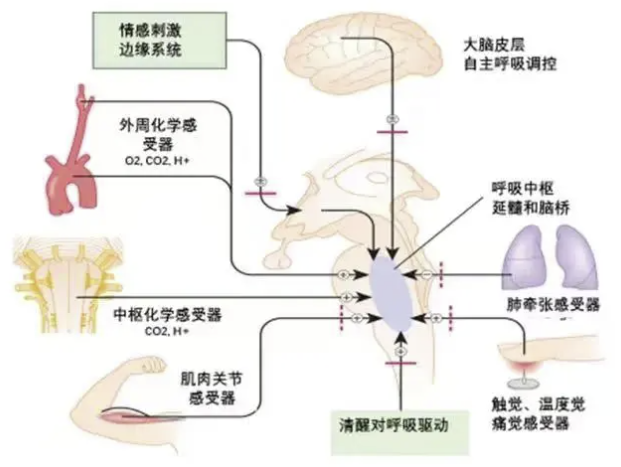
通气调控系统:在清醒状态下,通气由多重机制(包括中枢及外周化学感受器、机械感受器、行为性控制等)协同调控,代偿能力强,可维持血气相对稳定。但在睡眠(尤其卧位)时,生理状态改变导致调控能力下降:
通气效应器功能障碍:卧位时膈肌及肋间肌活动受限,出现限制性通气不足,潮气量降低,导致低氧血症。长期慢性缺氧可致化学感受器对低氧及高碳酸血症敏感性下降,调定点上移,基础血氧饱和度降低。
睡眠期调控恶化:睡眠中清醒刺激消失,通气转为以化学调控为主导。由于线粒体脑肌病累及神经系统:MELAS患者伴有乳酸酸中毒,乳酸酸中毒可导致GABA在突触间隙累积,导致化学感受器敏感性降低;脑干呼吸中枢神经元功能受损,呼吸驱动信号生成减弱;呼吸调控调定点上移,对低氧及高碳酸血症的通气反应显著钝化[4]。
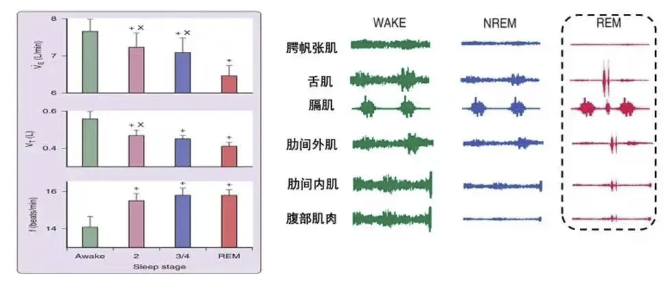
上气道稳定性受损:吸气相咽腔负压增大,而疾病所致咽部开大肌等上气道扩张肌肌力减弱,易诱发上气道塌陷,导致阻塞性睡眠呼吸暂停低通气低通气综合征(OSA)。
膈肌功能不全:作为主要吸气肌,膈肌在其他呼吸肌受累时代偿性承担更多负荷。线粒体功能障碍导致其肌力下降、易疲劳,收缩效能降低,致使功能残气量(FRC)增加,有效肺泡通气量减少。FRC降低减弱了肺对气道的径向牵引力(气管拖拽力),进一步减小上气道管径,加重气流受限,促成或加剧OSA。
3. 临床干预与病理联系
患者经夜间无创正压通气(NPPV,常用BPAP S/T模式)治疗后,慢性缺氧改善,化学感受器敏感性可部分恢复,坐立位血氧饱和度(93%)得以维持。这印证了其低氧血症主因源于睡眠期及卧位下通气调控与效应器的多重障碍,而非肺实质病变。
参考文献
1. PavlakisSG, PhillipsPC, DiMauroS, et al. Mitochondrial myopathy, encephalopathy, lactic acidosis, and strokelike episodes: a distinctive clinical syndrome[J]. Ann Neurol, 1984, 16(4): 481-488. DOI: 10.1002/ana.410160409.
2. 中华医学会神经病学分会. 中国神经系统线粒体病的诊治指南 [J].
3. 中华神经科杂志,2015,48(12):1045-1051.
4. Hypoxic ventilatory depression in a patient with mitochondrial myopathy, encephalopathy, lactic acidosis and stroke-like episodes. Osanai, S; Takahashi, T; Enomoto, H; Satoh, N,eta;RESPIROLOGY. 2001-06-01;6(2):163-6.
专家介绍

吕云辉
云南省第一人民医院睡眠医学科主任;主任医师,教授;中国睡眠研究会常务理事;中国老年医学会睡眠分会副会长;中国医师协会睡眠医学专业委员会常务委员;中国睡眠研究会睡眠医学发展工作委员会主任委员;云南省医师协会睡眠医学专业委员会主任委员。

邱敬满
云南省第一人民医院睡眠医学科主治医师,硕士研究生,中国睡眠研究会青年委员,中国极端环境与睡眠专委会委员,中国罕见病联盟发作性睡病专业委员会委员。从事睡眠医学临床、教学、科研工作10余年。参加国家自然科学基金项目1项,省级项目基金1项。发表SCI文章2篇。

刘畅
昆明医科大学 精神医学硕士,云南省第一人民医院睡眠医学中心主治医师,中国睡眠研究会青年委员;参与编写睡眠技术规范化培训教程;擅长抑郁障碍、焦虑障碍,失眠,发作性睡病,睡眠呼吸障碍等各类睡眠相关疾病的诊治研究。
* 文章仅供医疗卫生相关从业者阅读参考
本文完
采写编辑:冬雪凝;责编:Jerry