

 分享
分享
摘要
患者男性,16岁,入院前1年反复气胸、咯血、呼吸困难,于外院先后行5次胸腔闭式引流术,胸部CT示胸膜下肺大疱、右下肺团片状实变伴厚壁空洞形成,体检双手掌指关节、近端指关节、远端指关节可见过度活动,左侧踝部可见最大径1.5 cm淤血斑,基因检测结果显示COL3A1 c1816-2A>G剪切突变阳性。最终诊断为血管型Ehlers-Danlos 综合征。
患者男,16岁,因「间断胸痛、气短1年余,间断咯血7周」于2021年6月16日入院。患者于1年多前无明显诱因突发左侧胸痛,呈针扎样疼痛,深呼吸或咳嗽时加重,伴气短,日常活动即出现,外院X线胸片示「左侧气胸」,给予吸氧治疗后复查X线胸片示「肺复张良好」,上述症状亦缓解。
7个月以来患者反复出现气胸,肺压缩30%~40%,先后行「全身麻醉胸腔镜下左肺上叶局部切除、右侧肺大疱切除术+胸膜固定术」「左侧胸腔闭式引流及胸膜粘连术」,手术切除肺组织病理示「硬化性肺细胞瘤」。术后肺复张良好。
7周前、20 d前分别咯鲜血、暗红色血块各1次,量约5~10 ml,无发热、咳嗽咳痰、胸痛、呼吸困难等不适,均就诊当地医院,胸部CT示「右肺下叶不规则厚壁空洞、右侧少量胸腔积液、左肺多发胸膜下肺大疱」(图1, 2, 3),先后给予美罗培南(1.0 g,2次/d)、卡络磺钠(80 mg,2次/d)静脉滴注治疗12、14 d,治疗后患者咯血缓解。
6 d前患者无诱因再次咯鲜血1口,量约5 ml,于我院就诊。
既往患者自年幼时起,四肢皮肤轻微损伤后易出现瘀斑,双侧踝部为著。神经性呕吐4个月,口服米氮平、氟西汀治疗有效。口服奥美拉唑过敏(药疹)。青霉素、头孢曲松钠皮试阳性。否认家族遗传病史。
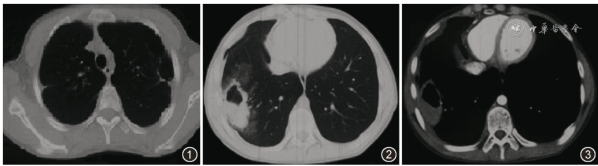
图1,2 2021年4月22日 胸部CT(增强)轴位肺窗可见双肺胸膜下多发肺大疱,左肺为著,右下肺胸膜下厚壁空洞病变,周围有少许磨玻璃样渗出影
图3 2021年4月22日 胸部CT(增强)轴位纵隔窗可见右下肺厚壁空洞病变内低密度区
入院体检:体温37.3 ℃,脉率78 次/min,呼吸频率20 次/min,体重指数(BMI)为17.3 kg/m²。消瘦体型,无特殊病容,皮肤菲薄,呈半透明,胸部及腹部皮肤浅表静脉明显可见,两侧腋前线第4肋间、肩胛线第6肋间、腋中线第7肋间均可见明显增生手术瘢痕。左侧踝部可见最大径1.5 cm瘀斑(图4)。双肺叩诊呈清音,呼吸音清,未闻及明显干湿性啰音和胸膜摩擦音。心脏和腹部体检未见明显异常。双下肢无水肿,未见静脉曲张。双手掌指关节、近端指关节、远端指关节过度活动(图5)。
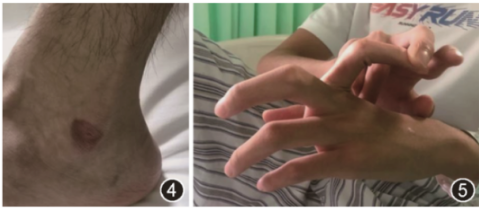
图4 左踝部皮肤轻微碰伤后瘀斑
图5 左侧掌指关节、指间关节过度活动
入院初步诊断:自发性气胸,肺脓肿?
实验室检查:血常规、尿常规、凝血指标、D-二聚体、肝肾功能均正常。血清抗核抗体、抗干燥综合征A抗体、抗干燥综合征B抗体、抗Sm抗体、抗Jo-1抗体、抗双链DNA抗体、血中性粒细胞胞质抗体(ANCA)均阴性;血清类风湿因子未见异常。血清曲霉半乳甘露聚糖抗原检测(GM试验)、真菌β-1,3-D葡聚糖检测(G试验)、烟曲霉特异性免疫球蛋白E、结核特异性的淋巴细胞培养+干扰素测定(T-SPOT)均阴性。腹部超声示肝胆胰脾肾均未见异常。超声心动示心内结构大致正常,左心室射血分数70%。眼科检查示晶状体、色素膜及眼底均未见明显异常。我院病理科会诊外院手术切除肺组织病理切片,肺组织部分区域梗死伴出血,局部纤维化,周围肺泡腔内广泛含铁血黄素细胞聚集,未见明显肿瘤及血管炎病变。入院后给予头孢美唑(2.0 g,2次/d)静脉滴注抗感染,卡络磺钠(80 mg,2次/d)静脉滴注对症治疗,咯血好转。7 d后复查胸部增强CT可见右下肺团片状实变影,空洞消失,病变范围较前缩小(图6,7)。检测患者及父母Col3A1基因,结果显示患者chr2:1898 62060位置出现杂合剪切突变c.1816-2A>G,其父母未见此突变表达。
最终诊断
血管型Ehlers-Danlos 综合征(EDS)。
由于本病目前尚无特异性治疗,给予患者对症治疗,如卡络磺钠、云南白药胶囊止血治疗,并健康教育,嘱患者避免外伤,避免高强度活动、剧烈咳嗽等。院外随访8个月,患者间断咯血3次,均为痰中带血或鲜血块,每日数口,外院对症止血治疗有效,未再发生气胸。
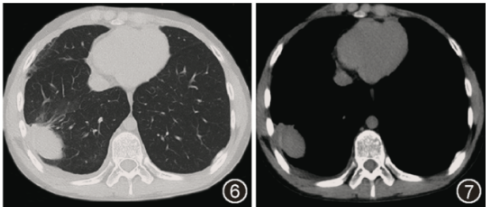
图6 2021年6月22日胸部CT(平扫)轴位肺窗示右下肺团片状实变影,空洞消失,较院外CT所示病变范围缩小,周围渗出影减少
图7 2021年6月22日胸部CT(平扫)轴位纵隔窗示右下肺团片实变影,内有低密度区
讨论
EDS是一组罕见的常染色体显性遗传疾病,又名弹力过度性皮肤、皮肤毛细管破裂,由丹麦皮肤科医生Ehlers和法国内科医生Henri-Alexance Danlos于1901年首先报道[1],其主要临床特征为皮肤超弹性、关节过度活动以及血管脆性增加。本病发病率为1/10 000~1/25 000[2],由于可能存在轻症患者未就医或被忽视,故真实发病率可能高于此数据。根据分子生物学诊断,可分为13种类型,即经典型、心脏-瓣膜型、血管型、脊柱侧后凸型、关节松弛型、皮肤脆裂型、脆性角膜综合征、脊椎变异型、肌肉挛缩型、肌病型。其中,血管型(Ⅳ型)最少见,仅占全部病例的4%,主要因Col3A1基因(编码Ⅲ型胶原蛋白)变异致病[3, 4]。本型EDS可出现极其严重的并发症,如动脉破裂、肠道或子宫破裂穿孔等,其他表现包括皮肤薄而透明、易于损伤淤血、先天性髋关节脱位、小关节过度运动以及颈动脉海绵窦瘘[4]。肺部表现包括空洞病变、气囊样病变、纤维结节、肺出血等,气胸或血气胸为本型区别于其他类型EDS的临床特征表现,可为首发就诊原因,发生率为16%左右[4]。与Birt-Hogg-Dube综合征中肺气囊病变多分布于下肺(隆突下)不同,本病分布无特征性优势[4, 5]。血管型EDS肺部病变,尤其气囊样改变,主要源于局部肺组织完整性差;纤维结节或炎性假瘤等可能源于肺血管或间质损伤后不充分修复,胶原Ⅰ型含量高于缺失的胶原Ⅲ型所致。
本例患者为青少年发病,临床特征如下:(1)反复发生严重的自发性气胸,伴间断咯血,即使外科胸膜粘连术、肺大疱切除术治疗效果仍欠佳;(2)小关节(双掌指关节、指间关节等)过度活动,皮肤轻度碰伤易于瘀青;(3)未发现心血管、眼部、肝脾肾等异常;(4)胸部CT表现为胸膜下肺大疱、右下肺团片状实变伴厚壁空洞形成,周围少许炎症渗出等;(5)无其他可导致肺部气囊样病变的疾病证据,如特殊感染(如结核/非结核分枝杆菌、曲霉)、结缔组织病、ANCA相关小血管炎、肺部肿瘤等相关检查均为阴性。检测患者及父母Col3A1基因,显示患者chr2:1898 62060位置出现杂合剪切突变c.1816-2A>G,其父母未见此突变表达,故考虑诊断为血管型EDS(散发性)。本病无特效疗法,主要以预防和治疗并发症为主,如对症止血治疗,尽可能避免动脉造影或介入治疗,减少胃肠镜检查操作,早期发现并处理动脉夹层等[4,6]。
本病需与其他可导致反复气胸的多系统受累的基因异常综合征鉴别,如Birt-Hogg-Dube综合征、结节性硬化症、马凡综合征等;亦需与其他可导致肺部空洞病变疾病鉴别,如特殊感染(肺结核、侵袭性肺曲霉病等)、系统性小血管炎等。
本病例中,高度提示可能为基因异常综合征的体征,如小关节过度活动、皮肤轻度碰伤后易于瘀青,在常规体检时容易被忽视漏检。因此,对于反复发生自发性气胸的患者,尤其是年轻患者,应注重关节、皮肤等全身系统体检,对于疑诊患者应尽早进行基因检测以明确诊断。
参考文献(略)
作者:李秋钰 张雨欣 宋祝 丁艳苓 朱红 孙永昌;单位:北京大学第三医院呼吸与危重症医学科
本文转载自订阅号「中华结核和呼吸杂志」
原链接戳:【病例报告】表现为反复气胸、咯血的血管型Ehlers-Danlos 综合征1例
引用本文: 李秋钰, 张雨欣, 宋祝, 等. 表现为反复气胸、咯血的血管型Ehlers-Danlos 综合征1例 [J] . 中华结核和呼吸杂志, 2022, 45(6) : 577-579. DOI: 10.3760/cma.j.cn112147-20211209-00869.