
 分享
分享

摘要
原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)是一种罕见的以呼吸道黏液清除功能障碍为主要表现的遗传性运动型纤毛疾病。近年来,PCD的临床及基础研究有较多进展,但目前中国PCD的早期诊断和治疗仍相对滞后。由此,由中华医学会呼吸病学分会呼吸遗传及罕见病学组(筹)、中国支气管扩张症临床诊治与研究联盟发起,在充分收集意见、查阅文献、线上和线下讨论和先前PCD中国专家共识的基础上,形成了新的PCD诊治中国专家共识。本共识收集了9个PCD相关的核心问题,内容包括PCD的临床表现、诊断、治疗以及患者管理和随访等。最终形成了9条推荐意见,以期提高中国PCD的诊断和治疗水平。
原发性纤毛运动障碍(primary ciliary dyskinesia,PCD)是一种罕见的以呼吸道黏液清除功能障碍为主要表现的遗传性运动型纤毛疾病。PCD最常见为常染色体隐性遗传模式,极少数为显性遗传或X连锁隐性遗传模式[1]。该疾病系基因突变导致纤毛结构和(或)功能异常,致使含有纤毛的组织器官发生功能障碍,严重危害人体健康。PCD全球发病率约为1∶7 554[2]。目前尚无中国PCD的流行病学数据。PCD的临床表现由受累的组织器官决定,不同基因突变会导致不同严重程度的PCD,但均有呼吸道纤毛功能受累的表现。PCD的常见临床表现包括新生儿呼吸窘迫、自幼反复发生的肺部感染、鼻窦炎、慢性中耳炎、支气管扩张症(简称支扩)、内脏反位(如右位心等)、不孕不育等。由于PCD临床表型差异较大,既往常易被误诊或漏诊。随着新技术的发展应用,包括透射电子显微镜(transmission electron microscope,TEM)、高速视频显微成像分析(high speed video microscopy analysis,HSVA)、免疫荧光显微技术(immunofluorescence microscopy,IF)、基因检测等[3, 4]均被逐步应用于PCD的综合诊断流程中,使PCD的诊疗水平飞速提高。2017年和2018年,欧洲呼吸学会(European Respiratory Society,ESR)、美国胸科学会(American Thoracic Society,ATS)先后发布了PCD的诊治指南[5, 6]。2020年,中国的呼吸界学者也首次制定发布了适合中国成人的《原发性纤毛运动障碍诊断与治疗中国专家共识》[7]。近年来,国际国内学者诸多PCD相关研究结果呈现,包括部分临床表型谱的新发现及细化、致病基因的迅速更新、诊断建议、诊断流程、患者管理等。鉴于此,由中华医学会呼吸病学分会呼吸遗传及罕见病学组(筹)、中国支气管扩张症临床诊治与研究联盟发起,基于近年来国内外最新研究进展,对2020年版中国专家共识进行了系统更新与修订。在广泛征求一线临床医务人员意见的基础上,通过问卷调查、专家访谈和文献查阅,最终确定了涵盖PCD的流行病学、临床表现、诊断、治疗以及患者管理和随访等方面的9个关键临床问题,并据此制订了《成人原发性纤毛运动障碍诊治专家共识(2025年版)》(简称“本共识”)。本次的修订更新了临床表型谱、诊断建议与诊断流程、患者随访及遗传咨询等方面的内容(表1),旨在提高临床医师对PCD的认识,促进PCD诊疗的科学性和规范性。

共识形成方法:(1)本共识由中华医学会呼吸病学分会呼吸遗传及罕见病学组(筹)、中国支气管扩张症临床诊治与研究联盟发起,组织呼吸与危重症医学、遗传学和循证医学等相关领域的专家共同完成。共识启动时间为2024年5月,定稿时间为2025年12月,并在国际实践指南注册平台(http://guidelines-registry.org/)进行了注册(PREPARE-2024CN1025);(2)目标人群和适用人群:本共识目标人群是PCD患者,适用人群包括各级医院呼吸与危重症医学科和遗传科医师;(3)证据检索:本共识的临床问题主要来源于对临床医务人员的调研、专家访谈以及相关文献经过整合后,并尽可能将问题解构为PICO格式(population,intervention,comparison,and outcome),并基于这些主题词进行检索。检索数据库包括PubMed、Cochrane Library、中国生物医学文献服务系统、万方数据库和中国知网数据库。检索时限为2024年11月,限定文献语言为中英文。(4)推荐意见形成过程:本共识中涉及诊断和治疗的部分内容,参考了分级的评估、制定和评价(grading of recommendation assessment,development and evaluation,GRADE)方法,进行证据质量和推荐强度的分级(表2)。证据质量分为“高、中、低、极低”4个等级,分别用A、B、C、D表示;推荐强度分为“强推荐/强反对、弱推荐/弱反对”2个级别,分别用1和2表示。共识初稿形成后,共识工作人员先后组织多次全体会议,对每个具体临床问题和推荐意见进行了充分讨论。所有推荐意见通过Delphi法进行投票表决。投票规则如下:对存在分歧的部分,推荐或反对某一干预措施至少需要获得50%的参与者认可,且持相反意见的参与者比例需低于20%,未满足此项标准将不产生推荐意见;一个推荐意见被列为强推荐而非弱推荐,需要得到至少70%的参与者认可。经过多轮讨论,最终确定本共识。所有参与共识制定的专家组成员声明不存在与本共识直接相关的利益冲突。

一、流行病学
PCD是一种表现为纤毛运动功能障碍的遗传性疾病,以常染色体隐性遗传形式最为常见,少数患者也出现罕见的常染色体显性遗传或X连锁遗传形式。PCD具有显著的遗传异质性,目前已鉴定的相关致病基因超过50个[8]。近年来,由于诊断设备的不断完善和普及,研究发现PCD常常被误诊或漏诊,最新的基于基因数据库的分析显示,PCD的患病率约为1∶7 554[2]。目前国内尚无大型PCD流行病学调查资料。
二、临床表现
PCD患者均有呼吸系统的临床表现[1],其他临床表现包括内脏反位、不孕不育、脑积水等[9]。
1. 呼吸系统:PCD患者呼吸道最典型的临床表现为慢性鼻窦炎和支扩[10]。患者通常自新生儿期出现鼻塞、流涕等慢性鼻炎相关症状,最终几乎所有患者发展为慢性鼻窦炎[11, 12]。成年PCD患者中,几乎100%有支扩,而在儿童中支扩的比例为56%~71%[13, 14]。其他较为常见的呼吸道临床表现还包括复发性急性中耳炎、慢性分泌性中耳炎和足月新生儿呼吸窘迫等[15, 16]。
2. 内脏反位:约50%的PCD患者会出现内脏反位,内脏反位的PCD患者合并先天性心脏病的风险较高(10%~12%)[17, 18, 19]。当患者同时出现支扩、慢性鼻窦炎、内脏反位三联征时,称为Kartagener综合征,可作为PCD临床诊断的提示[20]。
3. 不孕不育:男性PCD患者中约75.5%有不育的临床表现,主要表现为弱畸精子症(精子形态及运动能力异常)[21, 22]。PCD不会导致女性异位妊娠和流产,但约61.1%的女性PCD患者表现为不孕[21, 22]。
4. 脑积水:只有少数特定基因型的PCD患者有脑积水的临床表现[23],如FOXJ1基因突变患者均有症状较重的脑积水[24, 25],CCNO、MCIDAS基因突变患者易出现症状轻微或无症状的脑积水(约10%)[26],其他基因型的PCD患者脑积水的症状罕见。
5. 其他:少数PCD患者也会出现色素性视网膜炎、神经性耳聋、肾发育不良、多囊肾、脊柱侧弯、胸廓畸形、多指等临床表现[12, 13,20,27, 28, 29, 30]。
【问题1】哪些高危人群建议进行PCD筛查?
【推荐意见1】对既往存在支扩病史且同时具有以下任一病史的患者推荐进行PCD筛查:内脏反位;慢性鼻窦炎或鼻息肉;复发性或慢性中耳炎;不孕不育;近亲家系或同胞或直系亲属有PCD病史(1C)。
PCD相关诊断测试成本昂贵,需要复杂的设备和经验丰富的工作人员。因此,在临床工作中可以通过病史的询问及高危临床特征的识别,以简单确定哪些患者应该进行详细的筛查和诊断试验或转诊至PCD诊治中心。支扩是成人PCD患者最常见的临床表现,几乎所有的成人PCD患者均会出现[13,31, 32]。多项大型PCD队列研究显示,与其他病因所致支扩相比,以下临床特征,包括内脏反位、慢性鼻窦炎或鼻息肉、复发性或慢性中耳炎、不孕不育,以及有PCD家族史(包括近亲、同胞或直系亲属中有PCD患者)在PCD所致支扩患者中显著增高[33, 34, 35]。因此,上述病史的询问及高危临床特征的识别对于鉴别PCD具有较为重要的意义。
三、诊断
PCD患者临床和遗传异质性较强,目前没有单一的检测方法用于诊断所有PCD。PCD诊断需结合临床特征及多种辅助检查结果进行综合互补判断,目前临床上常用的PCD检查方法包括影像学检查、透射电子显微镜(TEM)、基因检测、经鼻呼出气一氧化氮测定(nNO)、高速视频显微镜分析(HSVA)和纤毛免疫荧光染色(IF)等。下文将分别介绍临床工作中各项辅助检查方法在PCD诊断中的应用与价值。鉴于不同于国外的国情,本次专家共识推荐了适合我国的PCD诊断分类和诊断流程(图1、表3)。
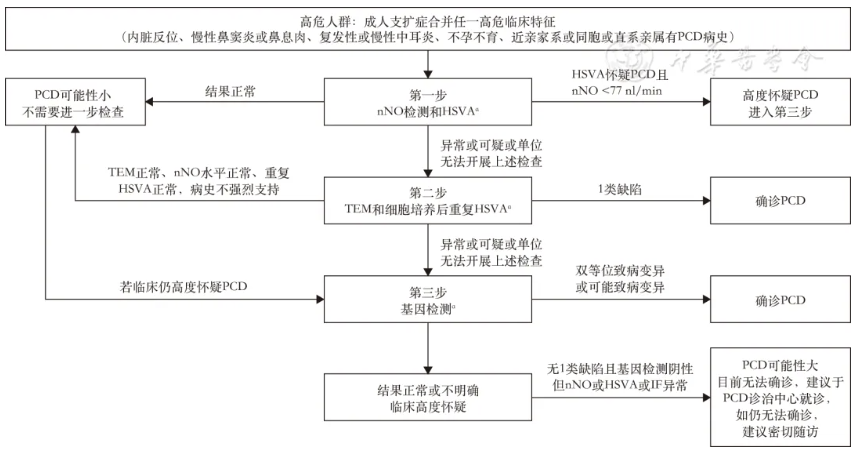
注:PCD为原发性纤毛运动障碍;nNO为经鼻呼出气一氧化氮;HSVA为高速视频显微镜分析;TEM为透射电子显微镜;IF为免疫荧光;aHSVA、TEM检测若单位无法进行,建议先进行基因检测或直接建议于PCD诊治中心就诊;现大多数单位可以借助测序公司进行基因检测,但结果解读建议联合遗传学专家共同进行
图1 成人PCD筛查和诊断流程图

【问题2】影像学检查在PCD诊断及管理中的价值如何?
【推荐意见2】对于上述PCD高危人群,推荐行影像学检查(包括胸部高分辨率CT、鼻窦CT、心脏彩超),有助于尽早发现提示PCD的临床特征,如支扩、鼻窦炎和内脏反位。同时,对于确诊PCD的患者推荐定期复查胸部高分辨率CT以评估肺部病变进展(1C)。
纤毛功能障碍累及多个器官,影像学检查应重点关注肺部、鼻窦、心脏和腹腔脏器等器官的表现。几乎所有的成人PCD胸部高分辨率CT(high resolution CT,HRCT)均显示出一定程度的支扩[13],病变主要分布在右中叶和左舌叶,其次为下叶,而上叶较少受累(图2)[13,32,35]。肺部的其他影像学特征还包括肺叶或肺段不张、黏液栓塞等,一般较少出现肺气肿和肺大疱。鼻窦CT可评估PCD患者是否存在鼻窦炎或鼻息肉。超声检查是评估心脏和腹腔脏器常用的检测手段,有助于评估PCD患者的内脏位置及是否合并先天性心脏病[17,36]。内脏位置的评估与判断对于患者以后可能的急性创伤评估或急诊手术时内脏器官的定位具有重要参考意义。
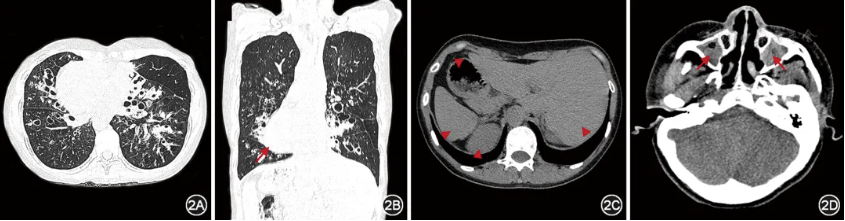
图2 典型原发性纤毛运动障碍患者CT所见 患者女,28岁,DNAH5双等位致病变异,胸部CT示右肺中叶、左舌叶及双肺下叶多发囊状支气管扩张,伴部分支气管管壁增厚、黏液栓塞,可见全内脏反位,长箭头示右位心,三角箭头示腹腔内脏全反位(胃、肝脏、脾脏、肾脏)(图2A~2C);鼻窦CT示双侧上颌窦黏膜增厚,窦腔见软组织密度灶填充,红色箭头示慢性鼻窦炎(图2D)
【问题3】透射电子显微镜(TEM)在PCD诊断中的价值如何?
【推荐意见3】对于临床高度怀疑PCD的患者,推荐使用TEM进行呼吸道纤毛超微结构检测。TEM作为可疑PCD患者的直接诊断依据,若检测出1类缺陷可直接诊断为PCD(1B),若检测出2类缺陷需结合其他检测结果综合判断。
既往对于TEM的检测及结果解读不规范,报告术语方面也缺乏统一共识,部分TEM结果存在漏诊或误诊的情况。近期,来自欧洲18个PCD中心的电镜专家发布了关于PCD电镜诊断标准的国际指南[37],规范了PCD患者TEM诊断中公认术语及国际通用超微结构分类体系。目前,TEM结果提示1类缺陷时可直接诊断为PCD(表4、图3),1类缺陷包括外动力蛋白臂(outer arm dynein,ODA)缺陷、内外动力蛋白臂(inner and outer arm dynein,ODA+IDA)缺陷、伴有内动力蛋白臂缺陷的微管组织紊乱(microtubular disorganisation,MTD)[37]。同时,TEM还可以识别某些非特异性或次要结构异常(2类缺陷,见表4)。当出现2类缺陷时,诊断PCD需结合患者临床病史和其他检测结果(如基因检测等)。尽管TEM诊断PCD特异度高,但敏感度较差[38, 39, 40],例如在微管连接蛋白、纤毛发生缺陷等相关基因突变的PCD患者中可能存在假阴性[9]。因此TEM检查结果正常时不能完全排除PCD,特别是对于具有典型临床病史的患者,需要结合其他结构与功能性检查综合评估。送检TEM样本组织时也应尽量避免炎症或感染较重的部位,以免对结果解读产生干扰。

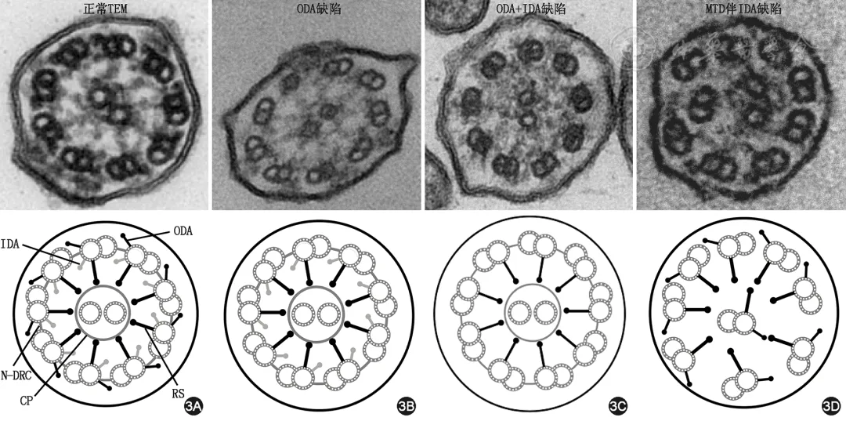
注:TEM为透射电子显微镜;ODA为外动力蛋白臂;IDA为内动力蛋白臂;CP为中央微管对;RS为辐射轴;N-DRC为连接蛋白-动力蛋白调控复合物;MTD为微管组织紊乱
图3 正常纤毛TEM及1类缺陷模式图 3A. 正常纤毛TEM及其模式图;3B. ODA缺陷及其模式图;3C. ODA+IDA缺陷及其模式图;3D. 伴有IDA缺陷的MTD及其模式图
【问题4】基因检测在PCD诊断中的价值和基因检测策略?
【推荐意见4】对于临床高度怀疑PCD的患者,推荐尽早使用全外显子组测序进行基因检测。全外显子测序作为可疑PCD患者的直接诊断依据,当出现已知PCD基因的双等位致病或可能致病变异时可直接诊断为PCD。在全外显子数据分析时,注意需包含拷贝数变异的分析(1C)。
基因检测有无创的优势,尤其对于TEM超微结构正常的PCD患者,全外显子组测序对其诊断尤为重要[1]。目前,已更新发现和证实50余个与PCD相关的双等位基因突变(见本刊网站附表),其中最常见的基因突变为DNAH5和DNAH11。检出已知PCD致病基因的双等位致病或可能致病变异可直接诊断为PCD。然而,临床工作中部分患者基因检测结果是意义未明的变异(variants of uncertain significance,VUS)或单等位基因杂合子变异,此时则需要进一步分析拷贝数变异(copy number variation,CNV)并结合功能和结构测试(HSVA、TEM、IF)来确定突变位点的致病性[41, 42]。此外,基因检测不能精确识别20%~30%的患者(此类患者可能存在目前未报道过的PCD新致病基因),其检测阴性仍不能完全排除PCD[34,43],需进行进一步功能试验。当临床高度怀疑PCD,而基因检测结果未发现已知的PCD致病基因变异时,建议进行家系(Trio)全外显子组测序,有助于发现新的PCD致病基因。
【问题5】经鼻呼出气一氧化氮测定(nNO)在PCD诊断中的价值如何?
【推荐意见5】推荐使用nNO作为可疑PCD患者的其他参考诊断依据,阈值为77 nl/min,低于阈值患者建议进一步行PCD确诊试验(1B)。
推荐可疑的PCD患者在初次进行肺功能检测时常规加入nNO检测,并且定期复查。nNO诊断PCD的阈值为<77 nl/min[检测结果单位nl/min与ppb换算方法:左右鼻孔平均浓度(ppb)×呼气平均流速(L/min)=最终nNO值(nl/min)][44]。需要注意的是,囊性纤维化、弥漫性泛细支气管炎、急性或慢性鼻窦炎、腺样体肥大、鼻息肉和上呼吸道感染等疾病均可使nNO降低[45, 46],出现假阳性。对于首次检测发现nNO值降低的患者应在至少2周后重复检测nNO,以确保nNO值降低不是继发于感染或其他可纠正的病因。研究报道PCD患者的部分基因突变(如RSPH1[47]、HYDIN[48]、CCDC103[49]、DNAH9[50]、DRC4[51]和NEK10[52]等)具有较高的nNO水平(超过上述阈值)。故nNO的结果不能作为单一诊断标准或者排除标准。当高度怀疑PCD,而该结果为阴性时,需要进行其他试验来确定诊断。
【问题6】高速视频显微镜分析(HSVA)在PCD诊断中的价值如何?
【推荐意见6】在有条件具备HSVA的临床机构中,推荐使用HSVA作为可疑PCD患者的其他参考诊断依据,阳性患者建议进一步行确诊试验(2C)。
HSVA是目前直观评估纤毛运动功能的重要测试方法,辅助PCD的诊断。正常纤毛的摆动呈波浪式,频率一般为12~15 Hz[53]。HSVA下PCD患者呼吸道纤毛显示摆动频率明显减低或摆动模式异常(包括完全静止纤毛、多数不运动伴残余运动的纤毛、摆动弯曲度小或振幅小的僵直运动的纤毛、运动功能亢进的纤毛等)[54]。研究报道HSVA诊断PCD的灵敏度和特异度可达到97%和95%~100%[55, 56],但目前为止,HSVA尚无统一的标准化流程和评价标准,且易受到囊性纤维化、继发感染或损害、环境、取材、设备等因素的影响,需要特定的设备和丰富的专业经验。同时,试验结果存在假阳性和假阴性可能,无法直接排除和确诊PCD。因此,建议有条件的PCD诊断中心将HSVA作为对于可疑PCD患者的其他参考诊断依据,定期复查,以识别需要进一步行确诊试验的PCD患者。此外,鼓励有条件的PCD诊断中心对可疑异常HSVA表现的患者进行纤毛上皮细胞气液交界培养以排除假阳性。
【问题7】免疫荧光(IF)在PCD诊断中的价值如何?
【推荐意见7】在有条件行免疫荧光的临床机构中,推荐使用IF作为可疑PCD患者的其他参考诊断依据,阳性患者建议进一步行确诊试验(2D)。
IF通过免疫反应识别纤毛的超微结构缺陷辅助PCD的诊断。通过IF可以识别几乎所有TEM检测到的超微结构异常,也可以识别部分TEM正常或轻微异常的病例。IF虽特异性强、灵敏度高[57, 58],但其缺点也很显著,如IF操作过程相对成本高、应用不广泛,而且使用特异性抗体特检测所有纤毛蛋白可行性低[59, 60],故目前尚未在临床上常规开展,仅推荐用于新基因鉴定的科研用途。另外,有研究表明,稳定期支扩患者的鼻上皮存在继发性纤毛结构异常,此类改变可能降低IF检测的特异性,结果呈阳性时并不一定反映原发性纤毛结构蛋白缺陷,从而导致假阳性[61]。
四、诊断分类及诊断流程
鉴于PCD的诊断较为复杂,虽诊断检查方法较多,但没有单一的检测方法用于诊断所有PCD。因此,目前采用多种互补性检查手段进行综合判断,推荐PCD的诊断分类及诊断流程如下:
根据ERS和ATS的PCD诊断指南,结合我国成人PCD临床诊治特点,将可疑PCD患者分为以下几类(表3):确诊PCD、PCD可能性大、PCD可能性小。PCD的筛查和诊断流程如图1所示。
五、治疗
PCD治疗目的包括延缓支扩疾病进展和减少急性加重,维持或改善肺功能,改善各系统的症状及患者的生活质量。
1. 支扩的内科治疗:由于PCD为罕见病,目前的大部分治疗方法缺乏循证医学证据支持。既往,PCD的支扩治疗主要参考支气管扩张症治疗指南及专家共识的治疗策略[5,7,62, 63]。急性加重期选用敏感抗生素(考虑是否经验性覆盖铜绿假单胞菌),并加强药物祛痰及物理性痰液引流。稳定期治疗以祛痰、气道廓清为主,可减少急性加重次数及改善患者肺功能。既往药物祛痰包括使用除痰剂、黏液调节剂、黏液溶解剂、黏液动力药等[64]。近期研究显示高渗盐水联合吸入上皮细胞钠离子通道(epithelial sodium channel,ENaC)抑制剂治疗,可使高渗盐水在呼吸道上皮中的初始和长期活性增加,维持足够的水合活性以恢复黏液水合作用,提高黏液清除率。CLEAN-PCD 2期临床研究证实ENaC抑制剂Idrevloride联合高渗盐水雾化治疗可有效改善PCD患者的肺功能并具有良好安全性[65]。
长期大环内酯类抗炎治疗在普通支扩治疗中较重要,但既往在PCD当中的研究较少。近期有研究显示在既往两年内因呼吸道感染或病情加重而接受过≥30 d抗生素治疗的PCD患者中,口服小剂量(250或500 mg/次,每周3次)阿奇霉素6个月后,PCD患者急性加重频率减半,痰液病原菌检出率显著降低[66]。因此,PCD患者的维持治疗可进行口服小剂量大环内酯类药物如阿奇霉素≥6个月,以减少支扩急性加重频率[66]。由于大环内酯类单药治疗会增加NTM和铜绿假单胞菌的耐药性。在开始长期大环内酯类治疗前,需明确患者有无活动性NTM感染、肝肾功能不全等情况,定期监测药物不良反应。
其他的治疗,包括支气管舒张剂、疫苗接种,以及并发症如咯血、慢性呼吸衰竭、肺动脉高压及肺心病等治疗,目前尚无相关循证医学依据,建议参考支气管扩张症专家共识中相关治疗策略。
2. 支扩的手术治疗:肺部外科切除术不作为PCD常规治疗推荐,仅限于出现严重局限性支气管扩张症伴反复感染,和(或)严重咯血内科及介入治疗均失败的患者。研究显示因PCD为广泛性气道受累疾病,肺叶(段)切除术通常不作为其常规治疗方法,其残存的气道清除功能将因术后疼痛和制动而进一步受限,患者术后短期内可有肺部疾病恶化的风险[10,67]。与非手术组相比,PCD肺叶切除术患者术后评估显示肺功能较差且持续下降,支气管扩张症再发率增加,提示不能从手术中获益[68],且痰液中铜绿假单胞菌的检出率更高[69]。
针对PCD终末期肺病可考虑行肺移植。迄今为止,研究报道全球PCD肺移植5年生存率在52%~70%[70, 71, 72],平均总生存期及无慢性肺异体移植功能障碍(chronic lung allograft dysfunction,CLAD)的生存期分别为5.9和5.2年,其中10年无CLAD期的PCD肺移植患者可达50%[71]。
3. 其他系统的治疗:PCD为多系统受累综合征,针对PCD其他系统的治疗,建议多学科会诊,包括PCD患者的鼻窦炎及中耳炎,通常推荐按照常规的鼻窦炎[73]及中耳炎[74]的治疗方案执行。确诊不孕不育的PCD患者建议生殖科专科就诊,尤其在专科医生的指导下选择合适的辅助生殖技术尤为重要[21, 22]。
4. 靶向治疗:鉴于既往PCD无对因治疗的方法,通常临床上针对不同受累脏器的具体病变进行对症治疗,但近年来,随着研究的进展,类似于囊性纤维化治疗的分子靶向药物的研发有了新的重大进展。基因靶向治疗可能是恢复PCD纤毛功能的希望所在。2024年6月,利用DNAI1 mRNA脂质纳米(雾化)的RCT1100药物疗法已被美国食品和药物管理局(FDA)授予PCD治疗的孤儿药认定[75],用于治疗由DNAI1基因致病突变引起的PCD。另外,PCD其他致病基因(如CCDC40、DNAH5)相关靶向治疗在开展的细胞及动物实验研究中也呈现了纤毛功能部分恢复[76, 77, 78]。相信在不久的未来,基因靶向治疗能为全世界更多的PCD患者带来新的治疗选择。另外,也推荐已明确基因突变的PCD患者积极参加基因靶向治疗相关临床试验。
六、患者管理
【问题8】PCD患者如何随访?
【推荐意见8】推荐成年PCD患者进行规律年度随访。随访内容主要包括肺部、鼻窦、耳的临床症状与功能评估,其他的评估内容包括疾病严重程度评估、免疫状态评估、生活质量及心理状态评估。另外,推荐育龄期PCD患者行相关不孕不育评估及随访(1C)。
对于症状严重的患者,应适当增加随访频率,每3~6个月随访1次[63,79]。如患者出现急性加重,应复查胸部HRCT、病原学检查,并进行合并症的评估。呼吸系统、耳、生殖系统是PCD的常见受累部位[7],需要定期进行随访。呼吸系统的随访内容包括呼吸系统症状[7]、胸部HRCT[13,80]、鼻窦CT[81]、肺功能(FEV1、FVC、FEV1/FVC、FeNO、nNO)及血氧饱和度[5, 6,81, 82, 83, 84]、呼吸道病原学[85]。耳的随访内容包括中耳炎症状及听力[81]。
疾病严重程度评估有利于对患者病情的综合判断,其主要评估内容包括:支气管扩张严重程度评估:支扩严重程度指数BSI[从年龄、体重指数(BMI)、FEV1%、过去2年住院次数、过去1年急性加重次数、mMRC评分、铜绿假单胞菌定植、其他病原微生物定植、支扩受累范围及类型多个方面进行评估]、E-FACED评分(既往1年是否因急性加重住院、FEV1%、年龄、铜绿假单胞菌定植、影像受累叶数、mMRC评分)、Reiff评分(影像学评估支扩严重程度)[86]、合并症评估(对PCD患者出现的严重合并症,应在每次随访时进行评估,评估内容包括咯血严重程度及原因,慢性呼吸衰竭及呼吸支持条件、肺动脉高压严重程度及干预手段)[63]。
评估PCD患者,尤其是反复、严重感染或机会性感染的PCD患者的免疫状态有利于疾病的管理[63]。免疫评估的内容包括:血常规(中性粒细胞计数、淋巴细胞计数)、免疫球蛋白(血清免疫球蛋白IgG、IgA、IgM)、必要时完善淋巴细胞亚群、补体、血清蛋白电泳等指标。
PCD通常表现为慢性病程,对患者的生活质量、心理状态进行规律评估有助于及早发现异常事件并及时处理。生活质量(quality of life,QoL)的评估内容包括支气管扩张症生活质量问卷(QoL-B)、支气管扩张症健康问卷(BHQ)、Leicester咳嗽生命质量问卷(LCQ),前两者为针对支气管扩张表现的疾病特异性生活质量评估工具,后者的评估内容基于咳嗽症状及咳嗽对日常生活的影响进行。近期系列研究开始使用焦虑-抑郁量表对PCD患者的心理状态进行评估[87]。
研究表明,与女性不孕(高达61%)相比,男性PCD患者不育发生率更高(高达83%)。然而,仅约一半的成年PCD患者(128例,48%)接受过生育相关随访。随访时平均年龄为30岁,且多发生在PCD诊断后平均10年[21,88]。
【问题9】遗传咨询如何帮助PCD患者家庭进行生育规划和决策?
【推荐意见9】为PCD患者家庭提供适当的遗传咨询可以帮助识别携带者,评估育龄人群子代患PCD的风险以帮助其作出恰当的生育决策(1D)。
遗传咨询通常包括以下几个步骤:(1)尽可能详尽地收集患者家系的人群资料和病历,包括家系成员的具体人数、性别、是否患病、临床表型及生育情况等;(2)进行必要的检查和遗传学检测,包括家系PCD致病基因变异筛查、先证者父母的PCD致病基因检测和产前诊断等;(3)进行子代患病风险的评估;(4)将检查和评估结果告知咨询者,并提供可能的选择;(5)对咨询者进行随访和扩大的家庭遗传咨询;(6)对咨询者进行心理评估[86,89]。
由于PCD致病基因众多,其中绝大多数PCD致病基因为常染色体隐性遗传模式,极少数致病基因为常染色体显性遗传或X连锁隐性遗传模式[9],因此父母双方携带的PCD致病变异所属基因是否一致及其遗传致病模式决定子代的患病概率。在父母双方携带同一PCD致病基因的变异,按孟德尔遗传定律:(1)若该基因的遗传致病模式为常染色体隐性遗传模式,当父母双方均为患者时,子代患病率为100%;当父母双方一方为患者时,一方为致病变异携带者时,子代患病率为50%;当父母双方均为携带者时,子代患病率为25%。(2)若该基因的遗传致病模式为常染色体显性遗传模式,当父母双方均为患者时,子代患病率为75%;当父母双方一方为患者时,子代患病率为50%。(3)若该基因的遗传致病模式为X连锁隐性遗传模式,当父母双方均为患者时,子代的患病率为100%;当父亲为患者时,母亲为变异携带者时,子代患病率为50%;当母亲为患者时,父亲为无突变的正常人时,子代若为男性则患病率为100%,子代若为女性则为正常表型的携带者。当子代存在患PCD的风险时,可以在孕前或孕期进行遗传咨询,以帮助PCD家系作出恰当的生育决策[84,90]。
中华结核和呼吸杂志《成人原发性纤毛运动障碍诊治专家共识(2025年版)》编写组
共识指导专家:
宋元林(复旦大学附属中山医院);瞿介明(上海交通大学医学院附属瑞金医院);徐金富
(同济大学附属同济医院)
方法学专家:
江梅(广州呼吸健康研究院广州医科大学附属第一医院);史静琤(中南大学)
专家组(按姓氏拼音排序):
白晶(广西医科大学第一附属医院);白久武(上海市肺科医院);毕晶(复旦大学附属中山医院);陈文慧(中日友好医院);陈智鸿(复旦大学附属中山医院);成孟瑜(山西白求恩医院);程哲(郑州大学第一附属医院);关伟杰(广州医科大学附属第一医院);郭婷(中南大学湘雅二医院);何萍(成都市第三人民医院);贺若曦(中南大学湘雅医院);胡晓文(中国科学技术大学附属第一医院(安徽省立医院));姜源(浙江大学附属儿童医院);李硕(天津医科大学总医院);李一诗(重庆医科大学附属第一医院);梁宗安(四川大学华西医院);刘念(湖南省人民医院);刘雅萍(中国医学科学院北京协和医院);罗红(中南大学湘雅二医院);马艳良(北京大学人民医院);欧阳若芸(中南大学湘雅二医院);秦铮(中国医科大学附属第一医院);沈宁(北京大学第三医院);谭志平(广东省深圳市坪山区中心医院);田欣伦(中国医学科学院北京协和医院);汪芹(中南大学湘雅二医院);王玺(北京大学第一医院);吴晓虹(浙江大学附属邵逸夫医院);夏丽霞(浙江大学医学院附属第二医院);谢敏(华中科技大学同济医学院附属同济医院);徐凯峰(中国医学科学院北京协和医院);杨欢(湖南省人民医院);袁雅冬(河北医科大学第二医院);张薇(哈尔滨医科大学附属第一医院);赵卉(山西医科大学附属第二医院);钟礼立(湖南省人民医院);周敏(上海交通大学医学院附属瑞金医院);曾奕明(福建医科大学附属第二医院)
文献质量评价组:
郭婷(中南大学湘雅二医院);卿洁(中南大学湘雅二医院);柳律(中南大学湘雅二医院);杨丹晖(中南大学湘雅二医院);雷诚(中南大学湘雅二医院);周湘林(中南大学湘雅二医院);杨彬艺(中南大学湘雅二医院);刘盈(中南大学湘雅二医院)
起草组:
郭婷(中南大学湘雅二医院);毕晶(复旦大学附属中山医院);李倬哲(复旦大学附属中山医院);唐薪竣(复旦大学附属中山医院);王文雯(复旦大学附属中山医院);卿洁(中南大学湘雅二医院);柳律(中南大学湘雅二医院);杨丹晖(中南大学湘雅二医院);雷诚(中南大学湘雅二医院);周湘林(中南大学湘雅二医院);杨彬艺(中南大学湘雅二医院);刘盈(中南大学湘雅二医院)
秘书:
郭婷(中南大学湘雅二医院);毕晶(复旦大学附属中山医院)
参考文献(略)
作者:中华医学会呼吸病学分会 中国支气管扩张症临床诊治与研究联盟;通信作者:罗红,中南大学湘雅二医院呼吸与危重症医学科 中南大学呼吸疾病研究所 长沙市人类纤毛疾病与转化医学科技创新中心 湖南省呼吸与危重症疾病临床医学研究中心 湖南省呼吸疾病诊疗中心;宋元林,复旦大学附属中山医院呼吸与危重症医学科;瞿介明,上海交通大学医学院附属瑞金医院呼吸与危重症医学科
引用本文:中华医学会呼吸病学分会, 中国支气管扩张症临床诊治与研究联盟. 成人原发性纤毛运动障碍诊治专家共识(2025年版)[J]. 中华结核和呼吸杂志, 2026, 49(2): 131-144. DOI: 10.3760/cma.j.cn112147-20250611-00322.
本文转载自订阅号“中华结核和呼吸杂志”
原链接戳:【诊疗方案】成人原发性纤毛运动障碍诊治专家共识(2025年版)
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry