
 分享
分享

间质性肺病(interstitial lung disease,ILD)是一类以肺间质炎症和纤维化为特征的疾病,可导致患者肺功能降低[1]。ILD包括多种类型,如特发性肺纤维化(idiopathic pulmonary fibrosis,IPF)、结缔组织疾病相关ILD和慢性超敏性肺炎等[1]。临床上一些少见的病因如胸腺瘤也可引起间质性肺病[2]。但由于胸腺瘤相关的间质性肺病病例较为罕见,临床上对该病认识不足,容易引起漏诊及误诊。我们报道了1例胸腺瘤相关自身免疫所致的间质性肺病,并对相关文献进行了分析与综述,旨在为该类间质性肺病的早期诊断和治疗提供参考。
1、病例资料
患者女性,66岁,因“咳嗽咳痰4年余,加重半年”入院。患者4年余前无明显诱因出现咳嗽、咳白痰,无胸闷气喘,季节交替时症状明显,受凉后加重,静脉用抗炎药后症状可缓解(具体不详)。近半年患者上述症状加重、痰转为黄色黏痰,量多。2024-05-04及2024-05-26于当地两家医院分别行胸部CT,均提示两肺纹理紊乱模糊,肺野透亮度增高,两肺斑片状、索条状高密度影,边缘模糊,支气管壁增厚,左肺下叶囊状透亮影,两肺门结构未见明显异常,气管、支气管通畅,纵隔内未见明显肿大淋巴结,两侧胸腔少量积液。双侧胸膜局部增厚。静脉予以红霉素、头孢菌素抗炎症状稍好转。1周前患者受凉后再发咳嗽咳痰,咳黄痰,伴活动后胸闷气喘,门诊查多排CT胸部平扫(图1):两肺间质性炎症;两肺支气管扩张;纵隔淋巴结稍大;两侧胸膜增厚;脂肪肝;肝囊肿可能。予以金荞麦片、头孢呋辛酯、左氧氟沙星等处理。现患者为寻求进一步诊治,收入我科。
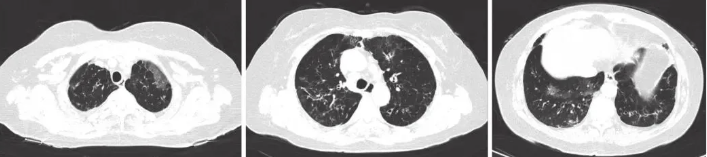
图1 患者2024年7月胸部CT像
两肺见斑片状、索条状高密度影,部分周围见囊状、柱状支气管影;纵隔淋巴结稍大;两侧胸膜增厚。
病程中,患者有咳嗽咳痰,无胸闷气喘,食纳睡眠尚可,大小便正常,近期体重无明显变化。既往:高血压7年余,平素服用降压药(具体不详)控制血压,血压控制尚可。2016年纵隔手术史,病理为胸腺瘤AB型(图2),2018年确诊肌无力,目前服用新斯的明片、硫唑嘌呤片控制。
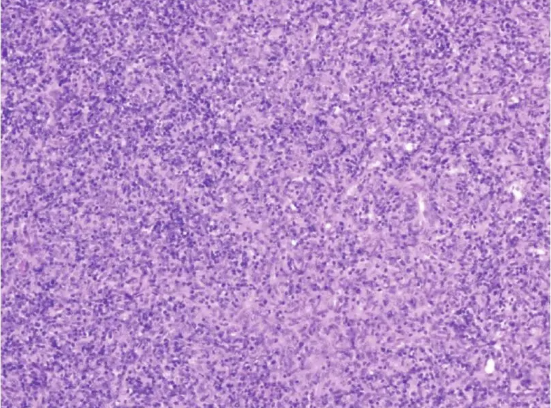
图2 纵隔肿物病理像(HE×100)
镜下所见符合AB型胸腺瘤。
查体:口唇发绀,无杵状指,双肺可闻及湿啰音。心脏无明显异常,双下肢无明显水肿。入院后查血气分析(FiO2:21%):pH 7.47,PaO2 45 mmHg(1 mmHg=0.133 kPa),PaCO2 40 mmHg,SO2 84%。血常规:白细胞计数10.011×109/L,嗜酸性粒细胞计数0.00×109/L,淋巴细胞计数0.97×109/L,淋巴细胞计数百分比9.7%。糖类抗原19-9:38.4 U/nL;糖类抗原72-4:7.75 U/mL;细胞角蛋白19片段:3.39 ng/nL。免疫球蛋白、类风湿因子、高敏肌钙蛋白T、血管紧张素转化酶等检查结果未见明显异常。淋巴细胞亚群绝对计数检测:淋巴细胞百分比5.59 %,B细胞百分比6.3 %,淋巴细胞计数604个/μL,总T细胞计数433个/μL,辅助/诱导细胞计数258个/μL,抑制性/细胞毒细胞计数164个/μL,B细胞计数38个/μL,NK细胞计数129个/μL,NKT细胞计数11个/μL。抗U1-小核糖核蛋白抗体(A-U1-snRNP)弱阳性,抗史密斯D1抗体(SmD1)弱阳性,抗组蛋白抗体(AHA)弱阳性。抗核抗体(ANA)均质型,1∶320。抗磷脂综合征、抗中性粒细胞胞浆抗体组套阴性,总IgE等检查结果未见明显异常。肺功能提示小气道功能障碍(操作过程中频繁咳嗽可能影响检查结果),肺一氧化碳弥散量60%~79.9%预计值,轻度弥散功能障碍。追溯病史,患者2016年曾行PET/CT检查,调取影像CT发现,肺部间质性改变(图3)。考虑胸腺瘤相关自身免疫所致间质性肺病,加用激素40 mg 3天后患者症状明显好转出院。
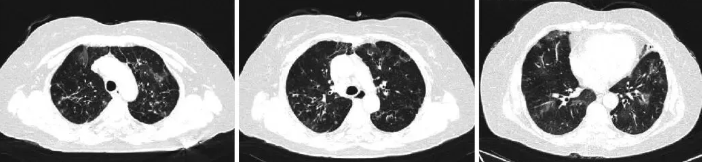
图3 患者2016年PET/CT肺部影像
前纵隔占位,两肺见斑片状、索条状高密度影,部分周围见囊状、柱状支气管影;纵隔淋巴结稍大;两侧胸膜增厚。
该患者出院后给予口服强的松30 mg qd,羟氯奎200 mg bid,半个月后(2024-08-07)在我院门诊复诊,药物减量为强的松25 mg qd,羟氯奎200 mg bid。2024-09-04患者复查胸部CT等后(图4)调整为强的松15 mg qd,羟氯奎200 mg bid。2024-10-09患者复诊后药物调整为强的松10 mg qd,羟氯奎200 mg bid。截至投稿时,该患者未再出现咳嗽、咳痰,指脉氧维持在98%~99%左右。
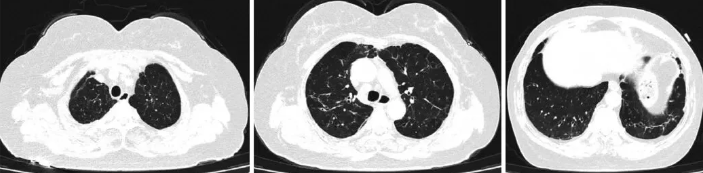
图4 患者2024年出院后胸部CT像(随访)
两肺见斑片状、索条状高密度影,部分周围见囊状、柱状支气管影;纵隔淋巴结稍大;两侧胸膜增厚;较2024-07-11部分吸收。
2、讨论
ILD是一组由200多种疾病组成的异质性疾病[3]。约30%~40%的ILD患者会发展为进展性肺纤维化,通常会导致呼吸衰竭,中位生存期约2.5~3.5年[4]。ILD的早期诊断和治疗一直是临床诊治的难点和重点。据文献报道,25%的ILD患者无法明确分类[5]。本例患者8年前发现胸腺瘤时即有肺部间质性改变,但因其无慢性呼吸道症状未予重视,4年后患者出现慢性呼吸道症状,本次由于合并急性感染,呼吸衰竭收治入院,最终通过临床检查、回顾病史及查阅文献,得到确诊。
该患者存在抗核抗体相关的部分抗体阳性,需明确是否合并结缔组织疾病(congenital tissue disease,CTD)相关的ILD(CTD-ILD)。然而,本例患者长达8年病程中未曾出现口干、眼干、反复腮腺肿大、光敏感、皮疹、指端溃疡或雷诺现象;无持续性炎性关节肿胀及超过1 h的晨僵;亦未表现出近端肌无力或肌酶升高等提示炎性肌病的临床表现。患者也未出现狼疮样皮肤损害、血尿蛋白或中枢神经系统受累等系统性硬化症、类风湿关节炎、皮肌炎/多发性肌炎、干燥综合征及系统性红斑狼疮(systemic lupus erythematosus,SLE)的特征性临床表现[6]。因此根据患者多次门诊与住院记录,临床表现并不支持CTD-ILD的诊断。患者实验室检查结果缺乏特异性。除抗核抗体(antinuclear antibody,ANA)均质型1∶320、抗U1-snRNP 弱阳性、抗SmD1弱阳性和抗组蛋白抗体弱阳性外,既往检查未发现抗Ro/SSA、La/SSB、CCP、RF、Scl-70、Jo-1、dsDNA及抗磷脂抗体等明显异常。ANA均质型1∶320提示核抗原反应,但ANA滴度与疾病严重程度并非线性关系;且健康人群、肿瘤患者或药物因素均也可致ANA阳性,须结合临床表现与特异性抗体综合分析[7]。抗U1-snRNP抗体虽与混合性结缔组织病及部分CTD重叠综合症相关,但单独弱阳性缺乏特异性;抗SmD1抗体对SLE诊断特异性高但敏感性低,在无临床特征不足以确诊SLE;抗组蛋白抗体常见于药物诱导狼疮,亦可在多种系统性疾病或肿瘤相关免疫激活中检出;而未分化结缔组织疾病虽可出现上述免疫指标异常,但诊断需症状持续至少1年,该患者明显不符合。综上所述,本例患者的多项抗体弱阳性更倾向于免疫紊乱而非明确的CTD诊断证据。与此同时,患者的胸部CT未见类风湿关节炎常伴的小气道病变或干燥综合征典型的囊性改变特征,也未出现系统性硬化症特征性的广泛基底外周纤维化伴食管扩张征象[8]。最后,需与具有自身免疫特征的间质性肺炎进行鉴别诊断。其诊断标准包括明确存在ILD(胸部高分辨率CT或外科肺活检证实),同时满足以下条件:(1)除外其他已知病因;(2)尚不能诊断某一确定的CTD;(3)至少具有临床表现、血清学表现及形态学表现的特征中的2个[9]。为进一步排除自身免疫特征的间质性肺炎,我们以“胸腺瘤”“胸腺瘤术后”及“自身免疫性疾病”为关键词的文献检索并进行了总结。
胸腺瘤是一种起源于胸腺上皮细胞的肿瘤,发病率低,但是作为成人最常见的纵隔肿块,占前纵隔肿块的50%[10]。胸腺瘤较易引起自身免疫性疾病(auto-immune disease,AID)和副肿瘤综合征(paraneoplastic syndrome,PNS)[11],56家机构联合研究表明38.8%的胸腺瘤患者继发AID和PNS,而在胸腺癌和神经内分泌胸腺肿瘤中,这两种疾病的发生率明显较低,分别为5.8%和3.6%。在整个队列中,重症肌无力是最突出的AID/PNS,占34.0%[10,12]。其他AID主要包括纯红细胞再生障碍性贫血、SLE、多发性肌炎和GOOD综合征。关于AID/PNS累及呼吸系统的文献报道极少[11]。Ferré等[13]报道,62名胸腺瘤患者中有13名患者存在肺部疾病(磨玻璃样阴影、支气管扩张症),这些患者均有慢性咳嗽或呼吸困难的症状。对这13名患者进一步研究发现,6名患者可检测出针对肺部特异性杀菌/通透性增加倍数B1(BPIFB1)和/或钾通道调节因子(KCNRG)的自身抗体。Maiolo等[2]则报道了1例胸腺瘤相关非特异性间质性肺炎的病例。3名合并有GOOD综合征的日本胸腺瘤患者均有弥漫性全细支气管炎,其共同点是HLA-B54阳性,但具体发病机制不明[14-15]。另有文献提示胸腺瘤患者也可合并结节病[16-17]。这些文献均提示胸腺瘤相关免疫失衡可靶向肺组织。而胸腺瘤切除后自身免疫性疾病仍可发生。有文献报道,21例胸腺瘤合并肾病的患者中,10例患者发生于胸腺瘤治疗后,间隔中位时间窗(108±83)个月,且部分病人ANA阳性[18]。其中的发生机制可能类似于本例报道,本例报道中患者ANA阳性,但并无CTD依据。本文报道的这例患者8年前切除胸腺瘤前肺部即已出现间质性病变,本次入院后影像学表现与其病情(呼吸衰竭)不符,常规抗感染效果不佳,及时加用激素后患者病情明显改善。综上可见,胸腺瘤引起的免疫异常可累及肺部,其表现形式多样,临床上容易误诊为CTD-ILD及具有自身免疫特征的ILD,临床上需充分重视该类患者的鉴别诊断及转归。
胸腺瘤相关自身免疫所致ILD机制未明。现有研究表明,胸腺是T细胞分化和成熟的场所,具有复杂的免疫功能,其对胸腺瘤发育和生长所引起的破坏性因素高度敏感。原始T细胞在胸腺中发育成有能力的免疫细胞,但在胸腺瘤中,T细胞不受控制,相互作用,相互攻击,成为影响人体内许多器官和组织的AID或PNS的触发因素。胸腺瘤患者发生自身免疫攻击的促成因素包括AIRE和MHCⅡ类表达水平被剥夺甚至缺乏,从而导致免疫耐受性降低以及调节性T细胞数量减少。自身免疫性T细胞和自身免疫调控机制的平衡被打破,从而促使AID的发生。同时,胸腺瘤细胞克隆的形成和扩增导致的胸腺结构紊乱亦会促进AID的发生[11]。也有研究发现胸腺瘤相关的自身免疫性疾病与其病理类型相关,AB型和B2型胸腺瘤由于能够产生更多的自身反应性T细胞更容易出现免疫功能异常[19]。肺泡上皮损伤或肺免疫反应失调可导致包括ILD在内的多种疾病[3]。ILD的常见危险因素包括吸烟、自身免疫异常、职业、环境和药物暴露等[20]。我们报道的这名患者其胸腺瘤病理类型为AB型,该型易引起机体免疫异常,造成上皮细胞损失,可能为该患者出现ILD的发病机制。
目前ILD的治疗主要包括抗肺纤维化治疗、激素治疗及免疫抑制剂治疗、肺移植治疗及肺康复等[21]。具体治疗方案的选择主要根据ILD的类型。除特发性肺纤维化、系统性硬化相关ILD及进展行肺纤维化外,其他种类的ILD的治疗主要为免疫调节治疗,过敏性肺炎主要为避免接触过敏原。该患者考虑为免疫异常相关的ILD,因此我们给予的治疗方案亦包含了激素及免疫抑制剂。
该患者肺部间质性改变在胸腺瘤确诊时即已存在,患者无明确职业、环境及药物暴露史,没有CTD的临床表现,肺部影像学特征也不符合CTD-ILD的典型表现,因此诊断诊断职业性、环境性、药物性及CTD-ILD依据不足。排除以上因素后,结合患者病史及胸部CT,考虑患者肺间质病变由胸腺瘤相关自身免疫机制导致。然而,本病例存在一定的局限性:患者长期未表现出口干、眼干或腮腺肿大等相关症状,初诊时未进行唇腺活检或眼科检查等客观评估,部分原因在于患者缺乏明确的侵入性检查指征。此外,由于患者随访方式受限于远程医疗条件,未能及时完成上述相关检查。我们已在近期随访中向患者明确建议进行腺体和眼科评估,如后续完成评估。胸腺瘤相关的肺部疾病罕见且机制复杂,临床工作中发现两种或多种病因不明疾病共存时,应细致分析其潜在关联。鉴于该类疾病的罕见性,加强长期临床随访尤为重要,以期为今后的临床治疗提供宝贵的参考依据。
利益冲突:本研究不涉及任何利益冲突。
参考文献略。
第一作者

黄文
医学博士,江苏省人民医院(南京医科大学第一附属医院)呼吸与危重症医学科副主任医师,南京医科大学讲师;研究方向聚焦于肺动脉高压、肺纤维化等;以第一/通讯作者发表多篇SCI论文。
通讯作者

张群
医学博士,江苏省人民医院(南京医科大学第一附属医院)呼吸与危重症医学科主治医师、副研究员,南京医科大学讲师。南京大学和美国匹兹堡大学联合培养医学博士。曾赴美国匹兹堡大学医学院联合培养及德国埃森大学鲁尔兰德临床医院(RUHRLANDKLINIK)进修;主持国家自然科学基金面上和青年项目2项,并获博士后基金及江苏省人才项目3项。研究方向聚焦于肺癌发生发展、肺纤维化及免疫微环境调控;以第一/通讯作者在Oncogene、Cell Death & Disease等发表多篇SCI论文。
引用本文:黄文, 何沂庭, 解卫平, 张群. 胸腺瘤相关自身免疫所致间质性肺病一例报道及文献复习. 中国呼吸与危重监护杂志, 2026, 25(1): 55-58. doi: 10.7507/1671-6205.202506088
本文转载自订阅号“中国呼吸与危重监护杂志”
原链接戳:【病例报告】胸腺瘤相关自身免疫所致间质性肺病一例报道及文献复习
* 文章仅供医疗卫生相关从业者阅读参考
本文完
责编:Jerry