

 分享
分享
摘要
本文报道1例有白化病的中年男性,主要临床表现为进行性呼吸困难,胸部高分辨率CT可见非特异性间质性肺炎,外科肺活检病理发现泡沫样、富含蜡样脂质脂褐素的Ⅱ型肺泡上皮细胞,局灶肺泡管及肺泡囊细支气管化生伴蜂窝肺形成,因不排除基因突变,行基因分析为HPS-1突变,确诊赫曼斯基-普德拉克综合征合并间质性肺疾病,经抗纤维化治疗但病情仍进展。
患者男,31岁,汉族,已婚,从事中医馆按摩师。因「反复咳嗽1年半余,活动后气促4月」于2019年3月入院。患者2018年1月无明显诱因出现干咳,2018年12月出现活动后气促,缓慢加重,走2层楼梯即可出现症状,病程中无关节痛、皮疹、口干眼干、雷诺现象,为求进一步明确诊断到我院就诊。
既往史:白化病,二级低视力残疾。个人史:无烟酒嗜好;从事中医馆盲人按摩,长期接触中草药材仓库,内有大量粉尘;家族史:姐姐白化病,但无肺部疾病病史。生育史:育1女,无白化病,配偶体健。
体格检查:脉搏78 次/min,呼吸18 次/min,血压126/76 mmHg(1 mmHg=0.133 kPa),指尖血氧饱和度94%(吸入氧浓度21%),皮肤、头发、睫毛呈白色,虹膜色素减退,杵状指,听诊双肺呼吸音粗,双下肺闻及吸气相Velcro啰音。
实验室检查:血常规、尿常规、粪便常规、凝血功能、心肌酶、pro-BNP、肝功能、肿瘤标志物(CEA、NSE、CA125、CA153)、抗CCP抗体、类风湿因子、抗核抗体定量、抗核抗体谱、抗血管炎抗体、抗心磷脂抗体、IgA、IgM、IgG均正常。血清涎液化糖链抗原-6(KL-6)为5256 U/ml(正常值<435 U/ml),血气分析(吸入氧浓度21%)pH值7.36,PaO2为62 mmHg,PaCO2为42 mmHg;6 min步行试验为423米,最低血氧饱和度为87%。肺功能检查为中度限制性通气障碍(FVC 占预计值为63.6%,FEV1 占预计值为67.4%,FEV1/FVC 90.57%),弥散功能中度下降(DLCO 占预计值为41.8%)。胸部高分辨率CT(HRCT)(图1~3):双肺弥漫磨玻璃影,细支气管扩张,近胸膜见网格影、网状线及牵拉性支气管扩张,双下肺容积缩小,叶间裂见增厚。
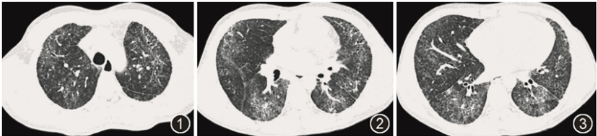
图1~3 患者胸部高分辨率CT(HRCT)显示,双肺弥漫磨玻璃影,细支气管扩张,近胸膜见网格影、网状线及牵拉性支气管扩张,双下肺容积缩小,叶间裂见增厚,考虑为间质性肺炎
第一轮多学科病例讨论(MDD):(1)影像科:HRCT表现双肺时相相对均一、分布相对对称的磨玻璃影伴细支气管扩张,双下肺可见网格影及牵拉性支气管扩张,考虑纤维化型非特异性间质性肺炎(NSIP);(2)呼吸内科:患者中年男性,慢性病程,长期接触中草药仓库;以活动后气促并进行性加重为表现;查体可见肺纤维体征;肺功能示限制性通气功能障碍伴弥散功能下降;胸部HRCT示纤维化型NISP。(3)风湿免疫科:患者无关节痛、皮疹、口干、眼干等症状,自身免疫疾病相关血清学阴性,不考虑结缔组织病导致的间质性肺疾病。经多学科讨论后,建议下一步完善外科肺活检取肺组织病理进一步诊断。
电视辅助胸腔镜外科肺活检(VATS-SLB),部位为左下肺前基底段(2 cm×1.5 cm×0.5 cm)、左上肺舌段(2.5 cm×2 cm×1.2 cm)。镜下:送检肺组织病变呈斑片状分布,肺泡腔内见多量泡沫样细胞及胆固醇结晶,Ⅱ型肺泡上皮细胞增生,其胞质呈显著泡沫样改变,个别细支气管鳞状上皮化生,肺泡管及肺泡囊细支气管化生,间质纤维化,可见蜂窝肺结构及新生的纤维母细胞灶,淋巴细胞浸润,小叶间隔增宽,见较多量血管及脂肪。泡沫样Ⅱ型肺泡上皮细胞强表达CK、SP-A、SP-B、TTF-1及NapsinA(图4, 5, 6),而CD163阴性;Galectin-3在间质纤维细胞、泡沫样肺泡上皮细胞及巨噬细胞均呈强阳性(图7, 8, 9)。六胺银、革兰、抗酸、PAS及D-PAS特殊染色均阴性。
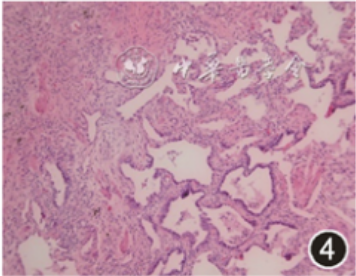
图4 患者肺组织病理学检查显示,病变呈斑片状分布,肺泡管及肺泡囊细支气管化生伴蜂窝肺形成,间质纤维化,可见新生的纤维母细胞灶 HE 低倍放大
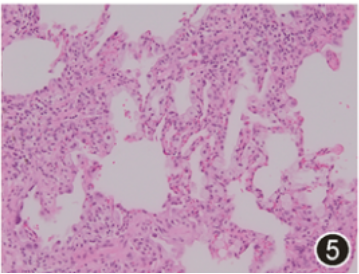
图5 间质性肺炎背景中见泡沫样、富含蜡样脂质脂褐素的Ⅱ型肺泡上皮细胞增生 HE 中倍放大
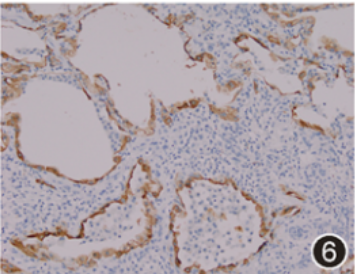
图6 CK在泡沫样Ⅱ型肺泡上皮细胞中呈强阳性,而肺泡腔内组织细胞阴性 EnVision法 中倍放大
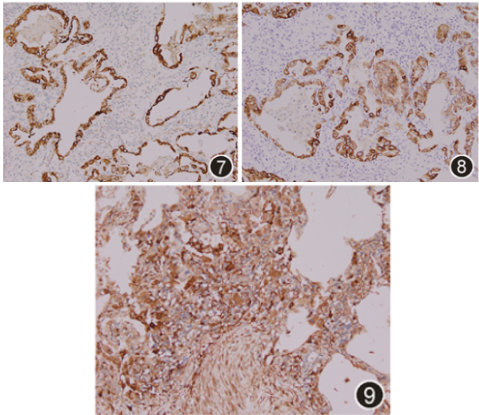
图7 SP-A在泡沫样Ⅱ型肺泡上皮细胞中呈强阳性,而肺泡腔内组织细胞阴性 EnVision法 中倍放大
图8 SP-B在泡沫样Ⅱ型肺泡上皮细胞中呈强阳性,而肺泡腔内组织细胞阴性 EnVision法 中倍放大
图9 Galectin-3在肺内纤维细胞、肺泡上皮细胞及巨噬细胞中均呈强阳性 EnVision法 中倍放大
第二轮多学科病例讨论(MDD):(1)病理科:肺病理考虑寻常型间质性肺炎(UIP),但是出现肺泡腔内见泡沫改变伴胆固醇结晶,不符合特发性肺纤维化(IPF)的病理。(2)呼吸内科:患者有白化病,肺组织出现肺泡上皮细胞大量泡沫样改变,不排除赫曼斯基-普德拉克综合征(Hermansky-Pudlak syndrome,HPS),需进一步做基因检测。
外周血基因检测发现患者HPS1基因序列上4号外显子187号位点发生纯合突变,碱基由G突变成T,最终导致HPS1基因表达终止(图10)。最终确诊HPS1型合并间质性肺疾病。患者接受3个月尼达尼布治疗(150 mg,2次/d),但活动后气促仍加重。2019年7月返院复查KL-6为5 880 U/ml,肺功能示中重度限制性通气障碍(FVC占预计值50.56%,FEV1 占预计值51.73%,FEV1/FVC 106.79%),弥散功能重度下降(DLCO 占预计值21.65%),胸部CT双下肺网格影、结节状、条索状磨玻璃影及密度增高影增多。因腹泻副作用,患者决定停用尼达尼布抗纤维化治疗。患者目前电话随访中,症状仍进行性加重。
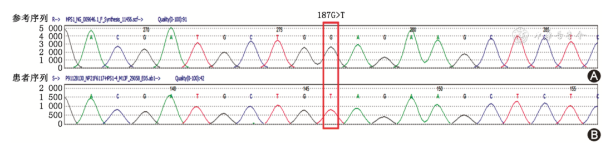
图10 患者基因检测结果显示4号外显示187号位点纯合突变
讨论
HPS是一种罕见的常染色体隐性遗传疾病,1959年由Hermansky和Pudlak最先报道,具有皮肤白化病、血小板功能障碍引起的出血、肺纤维化、肉芽肿性结肠炎等特征[1]。肺组织病理在HPS诊断过程中具有重要作用,但因HPS患者常出现血小板功能异常导致出血,所以经外科肺活检确诊的病例甚少。本例经VATS-SLB,除发现蜂窝肺、肺间质性改变外,还发现肺泡上皮细胞及巨噬细胞空泡样、富含蜡样脂质脂褐素的改变等HPS特征性表现,通过该病例可进一步提高病理科医生及呼吸内科医生对于该疾病认知。
HPS全球发病率仅为(1~2)/10万人,但是波多黎各岛的患病率高达约1/1 800,且接近1/22人携带突变的基因[2] 。至今已发现有10种HPS基因突变亚型,HPS基因表达的蛋白参与溶酶体相关细胞器(BLOC)复合物形成,包括BLOC-1、2、3和适配蛋白(AP)-3[3] 。HPS基因突变导致黑色素细胞或血小板的溶酶体相关细胞器(LORs)发生改变,黑色素细胞功能受损及血小板脱α颗粒造成聚集功能异常,所以出现眼皮肤白化病、虹膜色素退化及不同程度出血,细胞毒性T细胞功能异常亦可导致免疫缺陷[4, 5] 。
间质性肺疾病是成年HPS患者死亡的主要原因,在HPS-1、2、4型最为常见[6] ,HPS-1型和4型主要见于30岁以上患者,肺纤维程度重且预后差,HPS-2型主要见于儿童[7, 8] 。HPS-1和HPS-4是BLOC-3复合体的亚基,HPS-1和4基因突变导致BLOC-3复合体结构受损,肺泡上皮细胞及巨噬细胞功能异常,巨噬细胞和成纤维细胞异常激活,促进细胞外基质的积累和纤维化重塑最终导致肺纤维化[9] 。Nakatani等研究者[9] 从HPS死者肺部发现,Ⅱ型肺泡上皮细胞增生具泡沫肿胀及变形,以呼吸性细支气管炎为中心的淋巴细胞和组织细胞浸润,偶见缩窄性细支气管炎,不偏向于胸膜下分布的蜂窝样改变。病理中出现巨大板层小体变性提示肺表面活性物质的产生/分泌过程存在障碍,致细胞功能紊乱,诱发肺纤维化发生。本例HPS患者肺病理标本与朱翀等[10] 报道相似,肺部组织内可见泡沫样Ⅱ型肺泡上皮细胞,且细支气管扩张伴,免疫组化发现肺泡上皮细胞亦强阳性表达SP-A及SP-B,提示细胞内表面活性物质过度聚积;Galectin-3作为一种具有促纤维化作用的β-半乳糖苷结合凝集素,本例患者发现在肺内纤维细胞、肺泡上皮细胞及巨噬细胞中均有强表达Galectin-3。
HPS继发肺纤维化与IPF的胸部影像学具有相似性,都是双肺弥漫磨玻璃影、网格影及小叶间隔增厚,HPS疾病晚期也会出现蜂窝样改变,与IPF难以鉴别。但是,HPS继发间质性肺疾病的病灶主要沿着细支气管末端分布,双上肺网格影、牵拉性支气管扩张、蜂窝样改变亦多见,别于典型的IPF影像学改变[11] 。本例与徐铭蔚和张永明[12] 报道的病例相似,胸部HRCT都表现为双肺结节影、牵拉性支气管扩张,靠近胸膜处的网格影,影像学诊断为纤维化型非特异性间质性肺炎。但是,本例肺活检病理已经出现蜂窝样改变,与胸部影像学不相符合,说明患者病情更加严重,预后更差,未来可能会进展为双肺蜂窝样改变。此外,患者长期接触中草药仓库,存在霉菌孢子吸入,也可能是胸部HRCT出现广泛细支气管炎的因素,但是病理未见明确细支气管附近肉芽肿改变。
针对HPS合并肺间质病的治疗研究仍较少,一项纳入35例HPS合并肺间质病的前瞻性双盲随机对照研究,发现吡非尼酮不能延缓肺功能降低[13] 。本例患者3个月内FVC下降>10%,符合进展型纤维化型间质性肺疾病诊断标准,使用尼达尼布仍不能延缓肺功能下降,且使用尼达尼布需注意加重出血倾向。文献报道6例HPS患者,其中3例行肺移植,6年后无发现同种异体移植排斥或HPS致肺纤维化证据,总体认为肺移植是治疗HPS并肺纤维化有效手段[15] 。该患者达到肺移植指征,但因经济原因拒绝肺移植。
综上,HPS-1基因突变是HPS中最常见也是肺纤维化并发症最多见的一种亚型,白化病患者如出现无法解释的干咳、活动后气促,需做胸部HRCT了解有无间质性肺炎,并每年复查肺功能、胸部HRCT及6 min步行试验,胸部HRCT表现为双肺病灶以沿着细支气管末端分布磨玻璃影、牵拉性支气管扩张,肺组织病理具有特征性肺泡上皮细胞及肺内巨噬细胞泡沫样肿胀变形,伴细支气管扩张及淋巴细胞浸润。目前抗纤维化药物疗效尚不明确,肺移植可能是最有效的治疗手段。
参考文献(略)
作者:陈少华 郭炳鹏 刘春平 顾莹莹
单位:广州医科大学附属第一医院 广州呼吸健康研究院呼吸病理中心;广州医科大学附属第一医院 广州呼吸健康研究院 呼吸内科
本文转载自订阅号「中华结核和呼吸杂志」
原链接戳:【病例报告】赫曼斯基-普德拉克综合征继发间质性肺炎1例
引用本文: 陈少华, 郭炳鹏, 刘春平, 等. 赫曼斯基-普德拉克综合征继发间质性肺炎1例 [J] . 中华结核和呼吸杂志, 2022, 45(6) : 581-584. DOI: 10.3760/cma.j.cn112147-20211114-00803.