

 分享
分享

引言
本例患者以双上肢痛性红斑起病,进行性加重伴肌无力与咳嗽气喘,外院影像提示双肺间质性病变且抗感染治疗无效,病程呈快速进展趋势。入院后完善检查,确诊为“抗MDA5阳性皮肌炎合并快速进展型间质性肺炎(MDA5+DM/RP-ILD)”。
患者以“进行性皮疹+肌无力+快速进展的间质性肺炎”为核心表现,缺乏典型感染或肿瘤证据,早期极易误诊漏诊。在临床工作中,面对此类“皮疹合并肺部病变且治疗反应不佳”的复杂情况,需突破传统感染与常见结缔组织病的思维局限,将皮肤表现、肌力变化、影像动态与自身抗体谱置于系统性免疫性疾病的整体框架中串联分析,方能从错综复杂的临床表现中识别出潜在危重、进展迅猛的MDA5阳性皮肌炎相关间质性肺炎,为尽早实施针对性干预赢得关键时机。
皮肤痛性红斑、近端肌无力、肺间质病变……共同指向炎症性肌病?
卷宗1
基本信息:患者童某,41岁,因“皮肤痛性红斑2月,咳嗽咳痰1月”于2025年10月13日入住我院呼吸与危重症医学科。
讲述者:王晨
这位患者于2025年8月下旬无明显诱因出现皮肤红斑,以双上肢伸侧为主,部分呈硬结样突出皮面,伴局部疼痛,未重视,未就医诊治。至9月中旬,前述症状较前加重伴肌力下降,以四肢近端肌群为著,同时渐起咳嗽、气喘,偶有白痰,于当地诊所就诊,静滴抗生素治疗(具体不详)效果不佳,后咳喘及肌无力症状逐步加重,至当地县医院就诊,行胸部CT、颈椎MRI等检查,见双肺散在间质性病变,予抗感染等治疗后无明显改善,复查CT无改善,且肌无力症状加重,新发声音嘶哑,为系统性诊疗来我院就诊。
患者自患病以来,精神状态一般,体重无明显变化,食欲减退,二便如常,睡眠可。既往体健,否认慢性病史,否认烟酒不良嗜好史,否认遗传病史。个人史、婚育史及家族史均无特殊。
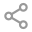
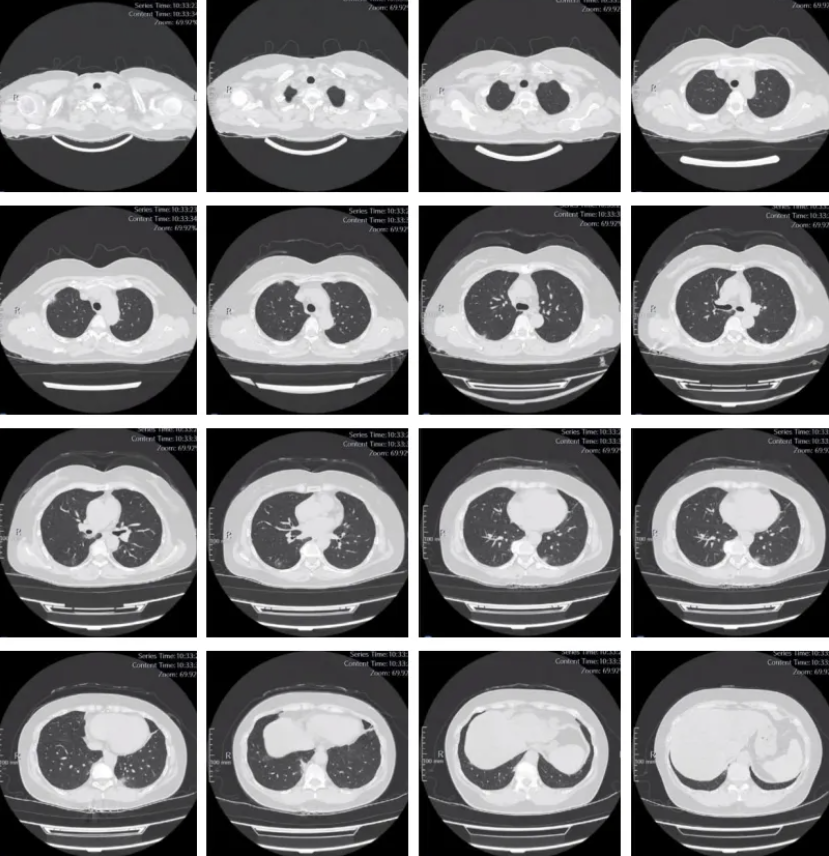
入院查体:T 36.2℃,P:86次/分,R:21 次/分,BP:122/70 mmHg。一般情况较差,搀扶步入病房,神清,稍萎,查体合作,被动体位。皮肤双上肢伸侧面及手肘可见皮肤角化加深,皮纹内散在破损及血痂;双手背侧面散在红色小丘疹,指尖皮肤角化伴脱屑。胸壁无明显异常,胸廓前后径正常,双下肺散在干啰音,以双下肺背侧为著。心脏及腹部查体大致正常;双下肢远端肌力5级,近端3级;双上肢远端5级,近端3级+;神经查体未见明显异常,双下肢无水肿。辅助检查情况:胸部CT(2025-9-24 外院):双肺多发炎症。
皮肤图片:
卷宗2
常规化验(2025-10-13):
血常规:白细胞:2.8×10^9/L、中性粒细胞%:65.2%、胞%:25.7%、单核细胞%:9.1%、嗜酸性粒细胞%:0.0%、嗜碱性粒细胞%:0.0%、中性粒细胞:1.83×10^9/L、淋巴细胞:0.72×10^9/L、单核胞:0.25×10^9/L、嗜酸性粒细胞:0.00×10^9/L、嗜碱性粒细胞:0.00×10^9/L、红细胞:3.07×10^12/L、血红蛋白:95g/L、红细胞积:28.30%、平均红细胞体积:92.2fL、平均血红蛋白浓度:336g/L、平红蛋白量:30.9pg、红细胞分布宽度CV:13.40%、血小板:127×10^9/L、均血小板体积:11.40fL、超敏C反应蛋白:18.02mg/L
生化:总胆红素:12.7umol/L、直接素:0.0umol/L、间接胆红素:12.70umol/L、总蛋白:56.0g/L、白蛋白:28.4g/L、球蛋白:28g/L、白球比:1.03、丙氨酸氨基转移门冬氨酸氨基转移酶:285U/L、葡萄糖:6.2mmol/L、尿素:4.1mmol/L、尿酸:181umol/L、肌酐:48umol/L
肌酶
肌酸激酶(CK):1970U/L、CK-MM:1956U/L
凝血功能
正常对照时间:11.2、凝血酶原时间:11.8S、INR:1.06、活化部血活酶时间:35.9S、凝血酶时间:17.9S、纤维蛋白原:3.28g/L、D-二聚体:3.12ug/ ml、FDP:9.1mg/L
结核相关: T-spot阴性,抗酸染色阴性。
炎症相关指标
血沉 87mm/h 降钙素原(PCT):0.0608ng/ ml、IL-6:41.1pg/mL
病毒检测
呼吸道病毒:人偏肺病毒、呼吸道腺病毒、呼吸道合胞病毒、副流感病毒核酸检测均为阴性;
流感病毒:甲型、乙型流感病毒抗原均为阴性。
免疫细胞亚群:指标及比例基本无异常。
免疫球蛋白
血清 IgG 14.10g/L,血清 IgA 2.25g/L,血清 IgM 0.34g/L,血清 IgE 211.00IU/ml,IgG4 1.14g/L。
传染病相关抗体:正常。
肿瘤标志物
癌胚抗原19.90ug/L、神经特异烯醇酶29.6ug/L、CA125:11.20U/ml、CA15-3:13.20U/ml、CA19-9 7.70U/ml、CYFRA211 3.53ug/l、铁蛋白 >2000.00ug/L、鳞状细胞癌相关抗原 0.5ng/ml。
自身免疫抗体:
抗核抗体:致密颗粒、抗体1:100:弱阳性、抗核抗体1:1000:阴性、抗核抗体1:10000:阴性、抗抗体:阴性、抗U1-RNP抗体:阴性、抗SSA抗体:阴性、抗SSB抗体:阴性、抗Jo-1抗体:阴性、抗Scl-70抗体:阴性、Ro52抗体:弱阳性、抗组蛋白AHA:阴性、抗核小体抗体:阴性、抗核糖体抗体:阴性、CENP B:阴性、肾小球基底膜抗体:0.107Units、MPO-ANCA:阴性、pANCA:阴性、PR3-ANCA:阴性、cANCA:阴性、抗双链DNA(定性):阴性、抗双链DNA(定量):9.70IU/ ml、抗环瓜氨酸肽抗体:0.606AU/ml
思考题:临床疑云,患者的肌力下降如何归因?
影像进展伴氧合骤降……肌炎与皮疹背后,究竟藏着什么致命危机?
讲述者:石昭泉
患者起病隐匿,以皮肤红斑和双上肢近端肌群肌力减退为首发症状,起初未系统就医,后上述症状逐步加重,出现双下肢近端肌力减退,入院前短时间内出现声音嘶哑伴吞咽困难,进展速度如此之快的肌力减退。
首先需要考虑的是神经系统疾病,重症肌无力通常表现为波动性,晨轻暮重,多累及眼外肌、延髓肌,且通常以外周肌群起病,逐步向心进展;那么是否需要考虑Lamber-Eaton综合征呢,该病多见于肿瘤相关的肌无力,虽然呈中心肌群开始的对称性肌无力,但通常与小细胞肺癌等具有神经内分泌功能的肿瘤相关,该患者影像学无典型表现,且实验室检验以炎症指标和肌酶升高为主要表现。
这时思路就相对清晰了,自身免疫介导的炎性肌肉损害,其首要考虑的疾病类型即特发性炎性疾病,在这个疾病门类里常见的疾病为皮肌炎、多发性肌炎、抗合成酶综合征等,但该患者自身免疫抗体均阴性,抗核抗体1:100阳性诊断价值极为有限。
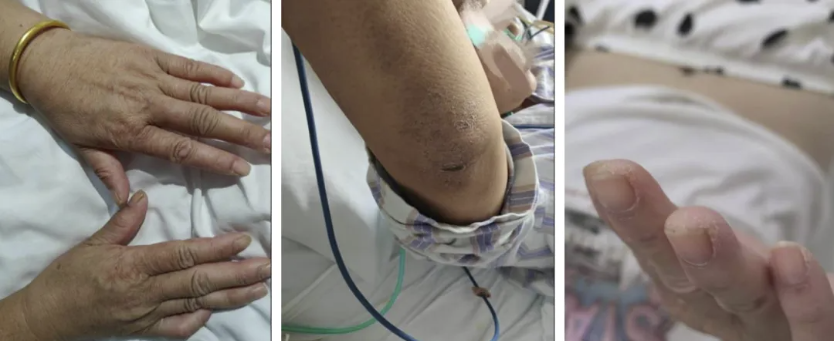
但很快患者的病情升级,影像学进展,氧和指数持续降低。入院后完善检查,患者血气分析提示低氧血症,复查胸部CT见双肺胸膜下病变范围进一步增大,部分呈间质样改变,结合患者“皮疹+肌损害+间质性肺病”的疾病模式,我们仍然考虑CTD-ILD,重点考虑肌炎相关的间质性肺炎,为进一步明确诊断,予外送肌炎特异性抗体谱25项抗体检测,在等待结果汇报的过程中患者干咳气喘症状逐渐加重,血气分析提示氧和指数逐渐降低。
同时,影像也陷入了重重迷雾。
但关键实验室指标为我们揭开了真相,原来,抗核抗体及ANCA阴性,肌炎特异性抗体暗藏玄机。
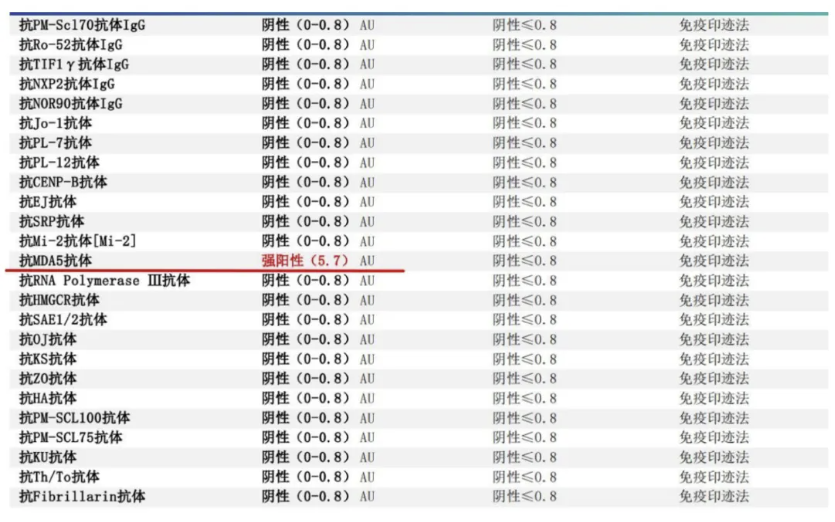
伴随着MDA5抗体阳性回报,诊断豁然开朗:
1、抗MDA5阳性皮肌炎(MDA5-DM)。
MDA5-DM是皮肌炎的一种亚型。目前,对于MDA5-DM尚无公认的诊断标准。MDA5-DM的临床诊断主要基于皮肌炎或IIMs的分类标准和MDA5抗体的检测,根据IIM广泛使用的2017年EULAR-ACR分级标准,成人皮肌炎可分为典型皮肌炎和无肌病性皮肌炎两类,2018年,欧洲神经肌肉中心将皮肌炎归为肌炎特异性抗体定义的亚组,包括MDA5-DM,其特征是Gottron征和向阳疹征,以及抗MDA5抗体阳性,但是部分患者皮疹不典型,仅是肺内受累严重,以及抗MDA5抗体阳性也可诊断为MDA5-DM。
该患者有较为典型的皮肤损害,双手指尖部分皮肤角质增厚伴脱屑样改变(技工手)、双手背侧及肘部、上肢背侧皮肤角化伴皮疹,部分呈破溃样表现(Gottron征)。且出现了非常明显的肌损害,肌电图等排除了神经系统损害,符合皮肌炎表现。在肌酶谱抗体强阳性检测结果回报后该患者MDA5-DM诊断清楚。
2、快速进展型间质性肺炎(RP-ILD)。
快速进展型间质性肺炎(rapidly progressive interstitial lung disease,简称RP-ILD)是指肺部间质性疾病在短时间内迅速恶化,导致呼吸功能急剧下降,需要紧急治疗的一种严重的肺部疾病,其病情恶化速度很快,通常在数周内呼吸功能急剧下降,甚至出现急性呼吸窘迫综合征,需要呼吸机辅助通气甚至肺移植。RP-ILD通常表现为广泛的肺部纤维化和肺泡炎症,预后通常较差。
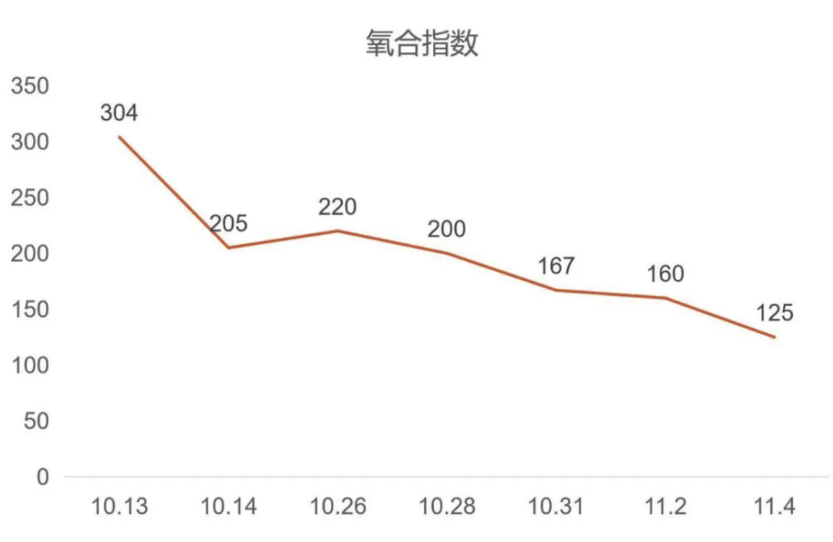
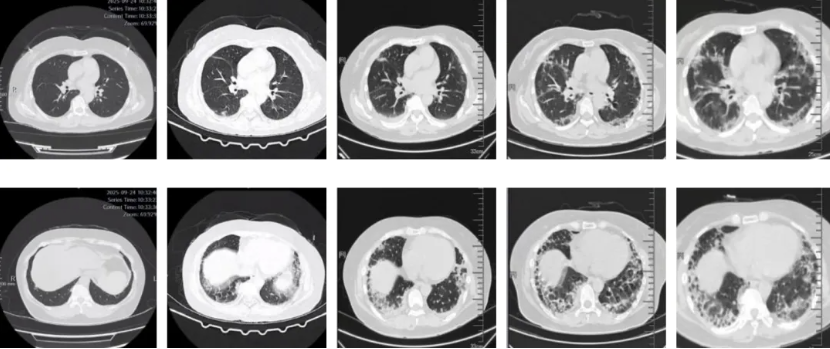
图:(从左至右)9-24、10-7、10-14、10-23、10-27
因此,该患者的诊断为:皮肌炎(MDA5阳性),快速进展型间质性肺炎,Ⅰ型呼吸衰竭。
患者入院后,予甲泼尼龙 80mg/d诱导治疗,抗体回报后上调至甲泼尼龙120mg/d联合环磷酰胺 0.4g/w*2w,并行静脉丙种球蛋白冲击治疗(0.4g/Kg*5d),排除实体瘤及结核、EBV等潜在治疗禁忌后,予托法替布 5mg 2/日联合FK506 3mg/d治疗。并行尼达尼布 150mg 口服 2/日抗纤维化治疗。
经治疗后,患者氧合改善不明显,转入RICU,更换HFNC支持,后续病情持续进展,经气管插管呼吸机辅助通气亦不能维持,需ECMO支持,最终因呼吸衰竭死亡。
抗MDA5抗体阳性皮肌炎合并RP-ILD:临床思维的核心引导
讲述者:王晨
特发性炎症性肌病(idiopathic inflammatory myopathies, IIM)是一种罕见的自身免疫性疾病,此病具有高度异质性,其主要临床表现为对称性的近端肌无力、肌酶升高,伴或不伴关节、皮肤、肺部以及心脏等其他组织器官受累的表现。与其他结缔组织病的自身抗体相似,肌炎自身抗体对IIM的诊断具有重要价值,主要包括肌炎相关性抗体(myositis associated autoantibody, MAA)以及肌炎特异性抗体(myositis specific autoantibody, MSA)两大类。MSA为致病性抗体,不同的MSA针对不同的自身抗原,常作为IIM的诊断、分型及预后评估的重要标志物。
2005年,日本学者首次发现了一种IIM的特异性抗体,此抗体与临床无肌病性皮肌炎(clinically amyopathic dermatomyositis, CADM)密切相关。在该抗体阳性的病例中,高达50%的患者发生快速进展型间质性肺病(rapidly progressive interstitial lung disease, RP-ILD),研究者将这种特异性抗体暂时命名为抗CADM-140抗体。进一步研究发现其抗原为黑色素瘤分化相关蛋白5(melanoma differentiation-associated protein 5, MDA5)所编码的蛋白质,因此将该抗体更名为抗MDA5抗体,该抗体对于抗MDA5抗体阳性皮肌炎具有非常高的诊断价值。
抗MDA5抗体阳性皮肌炎是一种特殊类型的皮肌炎,主要临床特征为抗MDA5抗体阳性、特征性的皮肤粘膜症状、较轻或不典型的肌肉受累和容易合并RP-ILD。此病在欧洲皮肌炎患者中的发病率仅有1.3%,而在亚洲皮肌炎患者中高达11%~21%,尤其以中国和日本多见。抗MDA5抗体阳性皮肌炎患者中,间质性肺病(interstitial lung disease, ILD)的发病率约为40%~100%, 而RP-ILD的发病率为40%~79%。
目前关于抗MDA5抗体阳性皮肌炎的发病机制仍不十分清楚,既往研究提示环境因素、遗传易感性、抗MDA5自身抗体和免疫细胞功能失调可能共同参与了该病的发病。
抗MDA5抗体阳性皮肌炎是一种全身性疾病,其临床特征具有高度的异质性,该病可以影响全身多个器官,但主要累及皮肤和黏膜、肌肉、关节、肺和心脏。皮肤粘膜主要表现为Gottron疹和/或Gottron征、向阳疹、V型疹、披肩征、皮肤溃疡、枪套征、甲周红斑、钙质沉着、技工手、雷诺现象等。肌肉关节症状主要表现为近端肌无力、肌痛、关节炎和关节痛。声音嘶哑、咽痛和吞咽困难也是抗MDA5抗体阳性皮肌炎较为常见的临床表现。
肺部是抗MDA5抗体阳性皮肌炎最常累及的脏器,往往影响患者的预后。在不同的抗MDA5抗体阳性皮肌炎人群中,ILD和RP-ILD的发生率存在显著差异。例如,在日本和东亚人群中,ILD的发生率达到82%~100%,RP-ILD的发生率达到60%~79%。而在高加索人群中,ILD的发生率为38%~67%,RP-ILD的发生率为20%~57%。
抗MDA5抗体阳性皮肌炎合并ILD,可以尝试通过高分辨率计算机断层扫描(HRCT),对ILD进行形态和病理类型的区分。主要包括机化性肺炎(organizing pneumonia, OP)、非特异性间质性肺炎(non-specific interstitial pneumonia, NSIP)以及OP与NSIP重叠,而寻常型间质性肺炎较为少见。需要注意的是,部分抗MDA5抗体阳性皮肌炎患者以ILD或RP-ILD为首发症状,没有典型的皮疹和肌肉受累。
抗MDA5抗体阳性皮肌炎相关ILD可能在HRCT上表现为严重的OP,直接导致弥漫性肺泡损伤,而无慢性肺部改变。下肺外侧带或沿支气管血管束分布的实变或磨玻璃影(ground glass opacities, GGO)是抗MDA5抗体阳性皮肌炎HRCT上的特征性表现,且与短期预后相关。通过动态监测HRCT的变化,对抗MDA5抗体阳性皮肌炎出现RP-ILD的早期诊断具有重要价值。HRCT显示进展为RP-ILD的特征性表现为在短期内从胸膜下或(和)小叶周围斑片状影迅速进展为广泛的实变影。
MDA5-DM合并RP-ILD的典型特征:
1.起病年龄:30–50岁。
2.起病速度:极快(数周内呼吸衰竭)。
3.肌炎程度:大多数无或仅有轻微肌痛、肌酶正常或轻度升高。
4.皮肤表现:几乎100%有特征性皮疹(溃疡性反Gottron征、机械手、皮肤溃疡)
5.实验室:铁蛋白升高(>1000–20000 ng/mL),LDH明显升高,抗MDA5滴度越高越危险。
6.肺活检或尸检:最常见弥漫性肺泡损伤+急性纤维素性机化性肺炎。
据现有研究证实的高危预后因素:
- 抗MDA5抗体高滴度
- 血清铁蛋白 >1000 ng/mL
- 起病时CT已有磨玻璃影或网格影
- LDH >400–500 U/L
- CRP明显升高(>10–20 mg/L)
- 外周血CD3+CD8+T细胞或NK细胞比例明显降低
我们的心得体会是:间质性肺病(ILD)是结缔组织病(CTD)患者常见的肺部并发症,临床工作中,如遇到肺间质性病变伴皮疹病例时,需首先考虑CTD-ILD可能。此外,快速进展性间质性肺病(RP-ILD)的诊断需要“稳、准、快”,这其中不应拘泥于抗核抗体和ANCA检测阴性,对于影像学快速变化、患者氧和情况持续恶化的病例,肌酶谱的检测意义重大。对于MDA5阳性-RP-ILD,强调早期联合治疗,目前早期大剂量糖皮质激素冲击联合免疫抑制剂治疗已形成共识,最新的研究也证实乌帕替尼等JAK通路相关的免疫抑制剂在该类疾病中展现出重要价值。
主任点评
这是1例较为典型的MDA5+DM/RP-ILD患者该患者有较为典型的“皮疹-肌损害-间质性肺炎”表现,MDA5 抗体作为关键诊断标志物,其检测对该病确诊至关重要。该病进展迅猛,预后极差,即使采用强化免疫抑制治疗,仍很难以逆转疾病进程。本病例提示,临床需提高对 MDA5 阳性ILD的认知,早期开展肌炎特异性抗体筛查,为及时干预争取时间。未来需进一步研究其发病机制,探索更有效的靶向治疗策略,以改善患者预后。
参考文献
[1] Hu H, Yang H, Liu Y, Yan B. Pathogenesis of Anti-melanoma Differentiation-Associated Gene 5 Antibody-Positive Dermatomyositis: A Concise Review With an Emphasis on Type I Interferon System. Front Med (Lausanne). 2022;8:833114.
[2] Chou JW, Lin YL, Cheng KS, Wu PY, Reanne Ju T. Dermatomyositis Induced by Hepatitis B Virus-related Hepatocellular Carcinoma: A Case Report and Review of the Literature. Intern Med. 2017;56(14):1831-1837.
[3] Bowles NE, Dubowitz V, Sewry CA, Archard LC. Dermatomyositis, polymyositis, and Coxsackie-B-virus infection. Lancet. 1987;1(8540):1004-1007.
[4] Mamyrova G, Rider LG, Haagenson L, Wong S, Brown KE. Parvovirus B19 and onset of juvenile dermatomyositis. JAMA. 2005;294(17):2170-2171.
[5] Sato S, Hoshino K, Satoh T, et al. RNA helicase encoded by melanoma differentiation-associated gene 5 is a major autoantigen in patients with clinically amyopathic dermatomyositis: Association with rapidly progressive interstitial lung disease. Arthritis Rheum. 2009;60(7):2193-2200.
[6] Vij R, Strek ME. Diagnosis and treatment of connective tissue disease-associated interstitial lung disease. Chest. 2013;143(3):814-824.
[7] Mukae H, Ishimoto H, Sakamoto N, et al. Clinical differences between interstitial lung disease associated with clinically amyopathic dermatomyositis and classic dermatomyositis. Chest. 2009;136(5):1341-1347.
[8] Lu X, Peng Q, Wang G. Anti-MDA5 antibody-positive dermatomyositis: pathogenesis and clinical progress. Nat Rev Rheumatol. 2024;20(1):48-62.
[9] Romero-Bueno F, Diaz Del Campo P, Trallero-Araguás E, et al. Recommendations for the treatment of anti-melanoma differentiation-associated gene 5-positive dermatomyositis-associated rapidly progressive interstitial lung disease. Semin Arthritis Rheum. 2020;50(4):776-790.
专家介绍

唐昊
上海长征医院(海军军医大学第二附属医院)呼吸与危重症医学科主任,医学博士,教授,主任医师,博士研究生导师;中华医学会呼吸病学分会青年学组副组长;上海市医学会呼吸病学分会委员兼秘书;上海市医学会呼吸病学分会哮喘学组副组长;上海市医师协会呼吸医师分会委员;入选上海市优秀学术带头人、上海市曙光计划、上海市浦江人才、上海市青年科技启明星、上海市卫生系统新优青等人才计划,承担4项国家自然科学基金,发表SCI论文30余篇。

石昭泉
海军军医大学附属长征医院呼吸与危重症医学科主任医师,教授,医学博士美国Lovelace呼吸研究所博士后,硕士研究生导师。上海市医学会呼吸专业委员会委员;上海市医学会变态反应专委会委员;上海市抗癌协会呼吸肿瘤内镜专业委员会常委;上海市康复医学会呼吸康复专业委员会委员;上海市康复工程研究会呼吸康复与医学工程专业委员会委员;国家自然科学基金、江苏、浙江、安徽省科委项目评审专家;教育部学位与研究生教育论文评审专家;上海市医疗事故鉴定专家组专家;上海市呼吸专业质控督查专家。从事呼吸专业30年,以哮喘、COPD的基础研究和临床防治及肺癌介入治疗为主要方向和特色。获国家、上海市各类基金10项,累计100余万元。以第一或通讯作者在国内外学术期刊发表论文近60余篇;主编或参与编写专著6部。

王晨
海军军医大学第二附属医院呼吸与危重症医学科医师,2022级PCCM学员。
* 文章仅供医疗卫生相关从业者阅读参考
本文完
采写编辑:冬雪凝;责编:Jerry